
Pourquoi diminuer le cholestérol ?
Bernard Bel 
 • Bookmarks: 1206
• Bookmarks: 1206
 • Comments: 2
• Comments: 2
 11138
11138

Le « mauvais cholestérol » serait-il la cause principale d’accident cardiovasculaire ou vasculaire cérébral ?
L’accident cardiovasculaire dont il est question ici est de ceux déclenchés par un syndrome coronarien aiguN1, autrement dit l’obstruction partielle ou totale d’artères coronaires, résultant de l’accumulation de plaque d’athéroscléroseN2. Cette obstruction entraîne un dysfonctionnement cardiaque et le plus souvent une nécrose partielle du muscle cardiaque privé d’oxygène (infarctus du myocardeN3). Le patient ressent une forte fatigue, une violente douleur thoracique et une sensation d’étouffement. Seul un traitement chirurgical en urgence permet d’éviter la mort : fibrinolyseN4, angioplastie coronaireN5 avec pose de stentsN6 ou pontage aorto-coronarienN7.
Il existe des variantes moins aigües de ce syndrome qui peuvent même passer inaperçues (sauf examen spécialisé) mais dont une réplique est potentiellement mortelle si aucune mesure préventive n’a été prise.
L’accident vasculaire cérébral (AVCN8) se présente sous deux formes gravissimes : hémorragiqueN9 qui résulte de la rupture d’un vaisseau dans le cerveau, ou ischémiqueN10 dans lequel un caillot sanguin ou un fragment de plaque d’athéroscléroseN2 bloque le passage du sang vers le cerveau — souvent au niveau d’une artère carotide. C’est seulement l’AVC ischémique (aussi appelé infarctus cérébral) qui s’apparente à un accident cardiovasculaire, avec des facteurs de risque comparables.
Sommaire
⇪ Athérosclérose
S’il reste difficile, en l’état actuel des connaissances, d’évaluer le risque d’AVC hémorragique, on peut déceler un risque d’AVC ischémique ou d’accident cardiovasculaire en constatant la formation de plaque d’athéroscléroseN2 dans les artères coronaires (proches du cœur) ou carotides (le long du cou). Cette détection met en œuvre des méthodes d’imagerie, plus ou moins invasives, qui vont de l’échographie DopplerN11 sur les vaisseaux du cou à l’angiographieN12 ou la scintigraphieN13 pour les coronaires. D’autres artères peuvent également être obstruées, avec pour conséquence des accidents vasculaires périphériques de moindre gravité : bras et jambes. Une détection indirecte de l’athérosclérose peut être faite à l’occasion d’un test de résistance à l’effortN14 qui révèle l’insuffisance d’apport d’oxygène aux muscles en mouvement.
Avant même ces tests, si l’on ressent un essoufflement anormal dans un effort habituel, des douleurs dans la poitrine, de la fatigue ou une paresthésieN15 dans les bras, etc., il est vivement recommandé de consulter un cardiologue.
La formation de plaque d’athérosclérose est directement associée à une augmentation des risques cardiovasculaire et d’AVC ischémique. Cette plaque crée un goulet d’étranglement local qui peut conduire à une interruption du flot sanguin si un caillot (thrombusN16) ou un fragment de plaque viennent l’obstruer. La stabilité ou la fragilité de la plaque est critique dans ce processus.
Une vidéo d’angioplastieN17 permet de se rendre compte de la complexité de la composition de cette plaque qui ne se réduit pas à un amas de susbtances graisseuses, contrairement aux affiches de certaines salles d’attente de cardiologues — offertes par des fabricants de statines… Cette composition est plutôt semblable à celle des fibromesN18 que l’on classe parmi les tumeurs bénignes.
Des études basées sur des techniques d’imagerie récente s’intéressent au mécanisme complexe d’évolution de la plaque d’athérosclérose, avec pour objectif d’évaluer le risque associé à sa fragilité. Une « macrocalcification » (de haute densité) augmenterait la stabilité, à l’inverse de microcalcifications liées à de la vascularisation, principalement entretenues par de l’inflammation (Nakahara T et al., 2017N19) :
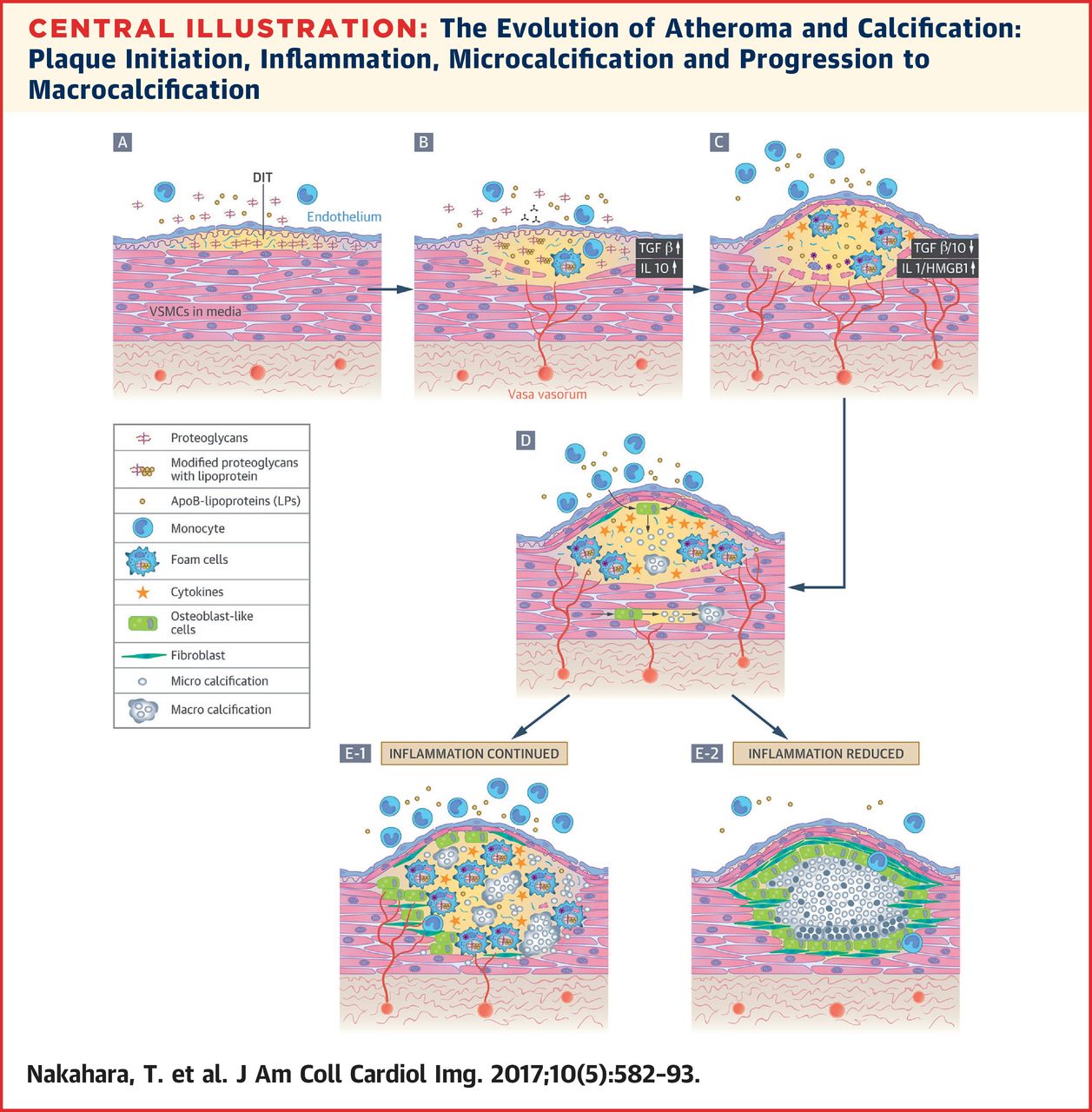
⇪ « Mauvais cholestérol(s) »
Lorsque, dans la vie courante, on parle de « mesurer le cholestérol », il s’agit en réalité de déterminer le taux sanguin de lipoprotéinesN20 porteuses, entre autres, de cholestérol. La distinction banale entre « bon » et « mauvais » cholestérol est celle, respectivement, entre les lipoprotéines de haute densité (HDLN21) et les lipoprotéines de basse densité (LDLN22). Pour un profil lipidique plus détaillé, il conviendrait d’ajouter, entre autres, les triglycéridesN23, ainsi que les lipoprotéines de très basse densité (VLDLN24), les lipoprotéines(a)N25 etc., qui n’apparaissent généralement pas dans les bilans sanguins mais peuvent influer sur la formation de plaque d’athérosclérose.
➡ L’abréviation LDL‑C désigne le cholestérol contenu dans les LDL, et HDL‑C celui contenu dans les HDL.
Une présentation claire du « bilan lipidique » a été publiée sur deux vidéos du neurologue Boris Dufournet (2023C4 ; 2022C3).
Des travaux en expérimentation animale ont montré que le rythme nycthéméralN26 modifiait à la fois le taux sanguin du LDL‑C et l’expression des gènes de ses récepteurs (Balasubramaniam S et al., 1994A3 ; Bray MS et al., 2011A8). Si un mécanisme comparable existe chez les humains, il pourrait expliquer certains aléas (et effets) de mesures du « bilan lipidique ».
L’expression « faire baisser le cholestérol » veut dire réduire le taux sanguin du LDL‑C sans diminuer celui du HDL‑C, ce dernier étant désigné comme « protecteur des artères » — une notion qui a été abandonnée récemment, voir Tricia Ward (2018A58).
Sachant que 20 % du cholestérol mis en circulation dans le sang est d’origine alimentaire et 80 % fabriqué par le foie, sous le contrôle d’un mécanisme de régulation, il existe en principe deux manières de maintenir un faible taux de cholestérol : intervenir sur le régime alimentaire ou/et utiliser des médicaments agissant sur le mécanisme de régulation. Les statinesN27 sont les plus répandus parmi ces médicaments, nous en parlons dans l’article Statines et médicaments anticholestérol.
⇪ Mesure ou calcul ?
Comment évalue-t-on le taux sanguin de LDL‑C ? En réalité, sa mesure directe est difficile et coûteuse. C’est pourquoi — sauf demande spécifique — les laboratoires se contentent de « calculer » ce taux en utilisant une équation proposée en 1972 par William Friedewald :
LDL (grammes par litre) = CT – HDL – TG / 5
où CT représente le taux de cholestérol total et TG celui des taux de triglycérides. Il est facile de vérifier, sur un bilan sanguin, la relation entre ces valeurs.
Cette méthode souffre de limitations qui peuvent conduire, par exemple, à une valeur excessive de LDL‑C lorsque le taux de triglycérides est faible, et donc à la prescription de statines sur une fausse base. De même, Friedewald et ses collègues ont reconnu que la formule n’était pas applicable lorsque TG dépassait 4 grammes par litre (soit 400 mg/dl).
Michael Eades a cité, dans The Arrow #117, un exemple de calcul qui avait conduit à une prescription (doublement !) inutile, tiré d’un article de Teh Y Wang et al. (2001A57) :
Notre patient est un homme de 63 ans en bonne santé qui a récemment subi un examen de routine qui a révélé les données de laboratoire suivantes provenant des laboratoires cliniques SmithKline Beecham de Seattle, Wash : CH [cholestérol total], 263 mg/dl ; HDL‑C, 85 mg/dl ; LDL‑C (calc.), 170 mg/dl ; et TG, 42 mg/dl. Les autres tests chimiques, les analyses hématologiques et les analyses d’urine n’ont rien révélé d’anormal. En raison des taux élevés de [cholestérol total] CH et de LDL‑C, son médecin de famille lui a prescrit de la pravastatine, à raison de 10 mg/jour. Avant de commencer à prendre son traitement, le patient a effectué un voyage d’affaires à Taïwan et, pendant son séjour à Taïwan, il a subi des examens de laboratoire similaires à l’hôpital chrétien de Sinlau.
Les résultats des tests effectués dans notre laboratoire [à Taïwan] étaient les suivants : CH, 262 mg/dl ; HDL‑C, 79 mg/dl, LDL‑C (calc.), 172 mg/dl, et TG, 55 mg/dl. [Autrement dit, identiques]
[…]
Nous avons examiné ses taux de TG anormalement bas et nous nous sommes demandés si cela n’aurait pas pour effet d’augmenter la valeur du LDL‑C (calculé) par rapport à la valeur réelle. Nous avons donc mesuré les valeurs de LDL‑C sur le même échantillon en utilisant la méthode directe et avons obtenu une valeur normale de 126 mg/dl ! Cette valeur a été confirmée par la méthode électrophorétique sur agarose (130 mg/dl) et a également été étayée par un taux normal d’apolipoprotéine B de 116 mg/dl (60–130 mg/dl).
Autrement dit, pour ce patient, la prescription d’une statine n’était pas indiquée — même en admettant « l’hypothèse du cholestérol » qui est revue ici de manière critique.
Une autre formule, plus précise lorsque les triglycérides sont élevés, a été proposée par une équipe iranienne (Ahmadi SA et al., 2008A2) :
LDL (grammes par litre) = CT/119 – HDL/110 + TG/190 – 0.38
⇪ Aperçu historique
Les chercheurs ont tenté d’établir (ou d’infirmer) un lien de causalité entre un taux élevé de « cholestérol » (ou de lipoprotéines de basse densité LDL) et la survenue d’accidents vasculaires associés à la formation de plaque d’athéroscléroseN2. C’est l’objet d’une controverse qui a débuté il y a une centaine d’années (travaux d’Alexander Ignatowski en 1908, cités par Tedgi A & Mallat Z, 2006A52) et s’est poursuivie autour de l’étude ENHANCE (2008N28).
Un récit détaillé de cette controverse est publié dans Cholestérol, mensonges et propagandes (de Lorgeril M, 2013B2 p. 53–84). Un autre récit bien documenté — humour anglais en prime ! — est l’introduction de l’excellent ouvrage The Clot Thickens (Kendrick M, 2021B6 p. 5–44). Le sujet est traité en détail par l’auteur anonyme A Midwestern Doctor (2025A1).
Avant la découverte des statines, les chercheurs ont étudié l’influence de l’équilibre nutritionnel sur (1) les taux de cholestérol sous diverses formes, (2) la survenue d’infarctus ou d’accidents vasculaires cérébraux, (3) la mortalité par accident cardiovasculaire et (4) la mortalité globale toutes causes confondues.
L’hypothèse du cholestérol comme cause principale de l’athérosclérose est issue de l’expérimentation animale sur des lapins (modèle de N. Anitschkov) et ultérieurement des rongeurs. La pertinence de ce modèle était douteuse puisque, par exemple, les pathologies d’artères coronaires n’existent pas chez les lapins, alors que ces animaux peuvent être victimes de lésions sur l’aorte, inconnues des humains. Les lésions artérielles, chez l’animal, sont disséminées, alors qu’elles sont localisées (plaques) chez les humains. Le modèle expérimental construit sur d’autres animaux, y compris les souris génétiquement modifiées, s’est révélé un échec pour l’étude des maladies artérielles (de Lorgeril M, 2013B2, p. 57–58).
L’étude épidémiologique de Framingham (1948N29) aux USA dans les années 1950, a tenté d’établir un lien de causalité entre la consommation de graisses qui augmenteraient le cholestérol et donc le risque d’infarctus, mais la corrélation n’a pas été observée. Gary Taubes (2007B7) signale que les chercheurs ont délibérément refusé de publier ce résultat, qui contredisait leur hypothèse initiale. Par la suite, ils ont mis au point une technique de centrifugation permettant d’isoler les lipoprotéines « HDL » et « LDL », les premières étant statistiquement associées à un moindre taux d’infarctus. C’est ainsi que s’est imposée la théorie du « bon » et du « mauvais » cholestérol. À la même époque, une étude semblable menée sur une population d’origine italienne dans la cité de Roseto en Pensylvanie, et publiée en 1964 dans le JAMA, montrait que l’association entre cholestérol et maladie cardiovasculaire n’était pas vérifiée (de Lorgeril M, 2013B2, p. 64–69).
L’étude de Framingham est aujourd’hui connue pour le calcul d’un index de risque d’accident cardiovasculaire à dix ansN30 fonction unique de l’âge, de l’index de masse corporelle, du tabagisme et des taux de HDL et LDL, sans tenir compte des habitudes alimentaires ni de la sédentarité, données occultées par les auteurs afin de désigner le cholestérol comme seul responsable de l’athérosclérose.
Dans la première moitié du 20e siècle, de nombreux pathologistes avaient observé, aux USA, l’absence de corrélation entre le taux sanguin de cholestérol et la sévérité de l’athérosclérose. Cette absence a notamment été constatée lors de l’autopsie de patients décédés. Le chirurgien Michael DeBakey, dont les observations portaient sur plus de 15 000 patients, déclarait en 1987 que les personnes dont le cholestérol est bas ont autant de risque que les autres d’être victimes d’athérosclérose (de Lorgeril M, 2013B2, p. 62).
Cette conclusion rejoignait celle de l’étude d’Amit Sachdeva et cal. (2009A48) portant sur 137 000 personnes hospitalisées pour une pathologie coronarienne : la moitié des patients avaient un taux de cholestérol LDL inférieur à 1.00 g/l, et la moyenne était de 1.049 ± 0.398 g/l, autrement dit dans la fourchette « normale »… L’étude de Vanessa S. Reddy et al. (2015A44) observant 115 492 patients hospitalisés pour un grave infarctus du myocarde, a reproduit sans surprise ce résultat, avec un taux moyen de LDL égal à 1.04 ± 0.38 g/l. Ses auteurs font état d’un “Lipid Paradox” car les taux les plus faibles de LDL étaient associés au plus haut risque de mortalité à l’hôpital, contrairement aux attentes.
Ayant étudié plus de 52 000 cas en Norvège (âgés de 20 à 74 ans) sur un suivi de 10 ans, Halfdan Petursson et ses collègues ont observé, surtout chez les femmes, une association inverse entre les taux de cholestérol et la mortalité globale, aussi bien que celle par accident cardiovasculaire. Ils déclaraient (2012A37) :
Si nos résultats sont généralisables, les recommandations cliniques et de santé publique concernant les « dangers » du cholestérol devraient être revues. C’est particulièrement vrai pour les femmes, chez qui un taux modérément élevé de cholestérol (selon le standard actuel) pourrait être non seulement sans danger mais plutôt bénéfique.
De Lorgeril (2015B3, p. 163) conclut :
Ces études suggèrent une fois de plus que la relation entre un taux de cholestérol élevé et une pathologie des coronaires est une falsification de l’histoire de la médecine. De même, la protection [contre cette pathologie] apportée par les statines présentée comme indubitable est aussi une falsification des méthodes scientifiques.
En 1957, une vaste étude épidémiologique a été lancée par Ancel KeysN31 : l’Étude des Sept PaysN32 pour mesurer l’influence du régime alimentaire sur la santé. Cette étude couvrait les populations des États-Unis, Finlande, Hollande, Italie, Grèce, Yougoslavie et Japon, recherchant entre autres des facteurs susceptibles de favoriser l’infarctus du myocarde : cholestérol, tabac, tension artérielle et indice de masse corporelle. Les résultats ont été publiés quinze ans plus tard et ont donné lieu à vingt ans de suivi. Les habitudes alimentaires, le tabac et la tension artérielle ont bien été identifiés comme facteurs de risque cardiovasculaire. Pour le cholestérol, les résultats font l’objet de critiques — voir Criticism sur la page WikipediaN32. Michel de Lorgeril écrit (2013B2, p. 70) :
Keys aurait pu tirer des conclusions très différentes. Par exemple, ce sont les graisses saturées qui indiquent le risque (et pas le cholestérol) ou bien qu’un ratio graisses saturées/monoinsaturées bas (rappelant la diète méditerranéenne) est protecteur. Mais il préféra rester fidèle à la théorie du cholestérol et concentrer son message presque exclusivement sur le cholestérol.
Par ailleurs, les disparités entre pays des associations entre cholestérol et infarctus posent le problème de la validité d’une telle association. Elle n’est pas significative pour les Japonais. L’étude est biaisée par le choix des pays. Par exemple, en incluant la France, la Suisse ou la Belgique, pays qui avaient un taux moins important de maladies cardiovasculaires malgré une plus forte consommation de graisses saturées et un taux plus élevé de cholestérol, la démonstration s’effondrait. C’est cette disparité que des épidémiologistes, à la fin des années 1980, ont désigné par « le paradoxe français » (French paradoxN33) mis en évidence par Serge RenaudN34 — voir le commentaire amusant de David M Diamond en vidéo (2017N35). De Lorgeril ajoute (2013B2 p. 73) :
Une autre erreur de Keys fut de se focaliser sur la mortalité cardiovasculaire au détriment des chiffres de la mortalité totale. Pourtant, ce qui nous intéresse le plus, ce n’est pas de savoir si en changeant d’alimentation nous pouvons échapper à une maladie cardiovasculaire, mais de savoir si nous évitons les maladies (quelles qu’elles soient) qui diminuent l’espérance de vie.
L’étude MRFIT (Multiple risk factor intervention trialA33) publiée en 1982 a mesuré l’effet de recommandations de vie saine sur 12 866 Américains, âgés de 35 à 57 ans et sélectionnés comme « haut risque » de par leur taux de cholestérol très élevé (environ 3 grammes par litre). Un groupe tiré au hasard (SI) a suivi les recommandations d’améliorer son alimentation et de cesser de fumer. Les deux groupes d’hommes ont été suivis pendant 7 ans. Les auteurs reconnaissent que le taux de mortalité cardiovasculaire avait diminué de manière non significative (de 19.3 à 17.9 ‰) dans le groupe (SI), tandis que le taux de mortalité totale n’avait diminué que de 41.2 à 40.4 ‰ dans ce même groupe. Ils concluent qu’une explication plausible, bien que nécessitant « une étude plus approfondie », serait que dans le groupe (SI) certaines personnes ont réellement bénéficié de l’arrêt du tabac et de la réduction du cholestérol, tandis que pour d’autres ces mêmes mesures auraient pu avoir un effet adverse au traitement de l’hypertension artérielle… De Lorgeril précise (2013B2 p. 75, 77, 78) :
En fait, les décès étaient même plus nombreux chez les hommes auxquels on avait conseillé d’arrêté de fumer, de suivre un régime anticholestérol, et de surveiller leur pression artérielle, comme si l’adoption d’un régime anticholestérol avait augmenté leur risque. […] Enfin, quand de jeunes analystes ont voulu vérifier si la relation observée dans MRFIT entre cholestérol et décès cardiaque était influencée par d’autres facteurs, ils eurent la surprise de constater que cette association était surtout décelable chez les fumeurs et presque absente chez les non-fumeurs. […] Cet aspect est capital car ces facteurs d’accompagnement sont probablement certaines des véritables causes des décès cardiaques. Je les appelle facteurs Z. Ces facteurs ne sont pas les seules causes de l’infarctus et des décès cardiaques, certes, mais ils agissent aussi sur le cholestérol et dès lors laissent croire que c’est lui le coupable.
Le Wall Street Journal titrait, le 6 octobre 1982 : « Infarctus : une hypothèse s’écroule. » (ibid.)
Michel de Lorgeril (2013B2, p. 81–84) présente enfin l’étude INTERHEART (Yusuf S. et al., 2004A60) dans laquelle les auteurs ont substitué à la mesure du cholestérol celle du rapport apolipoprotéine BN36 sur apolipoprotéine A1N37, affirmant que le rapport B/A1 est un équivalent du cholestérol, ce qui est faux parce que l’apolipoprotéine B est présente dans toutes les lipoprotéines (contrairement au cholestérol) et notamment les lipoprotéines riches en triglycérides. Cette apolipoprotéine B capture donc, en plus du risque associé au cholestérol, le risque associé aux triglycérides qui en est indépendant, au moins en grande partie (ibid.). D’autre part, les chercheurs ne se sont intéressés qu’aux cas d’infarctus non-mortels (environ 50 %). De Lorgeril souligne :
L’augmentation des triglycérides est souvent associée à des modes de vie ou des caractéristiques physiologiques tels que la sédentarité, le surpoids ou l’obésité, la résistance à l’insuline, ou d’autres syndromes métaboliques, certaines formes d’hypertension et même le tabac. La plupart de ces facteurs n’ont rien à voir avec le cholestérol. […] Le même raisonnement s’applique à l’apolipoprotéine A1.
L’étude de Wilkins, JT et al. (2016A59) confirme qu’une augmentation du taux sanguin des apolipoprotéines B N36 serait un marqueur de risque de survenue d’une maladie cardiovasculaire, et ce, de manière indépendante du niveau du LDL cholestérol (cité sur WikipediaN36).
Dans la suite du même chapitre (op.cit. p. 92–95), De Lorgeril commente trois études interventionnelles sur des patients ayant survécu à une première crise cardiaque : études d’Oslo (publiée en 1970), de Londres (1968) et de Sydney (1978). On répartissait les patients en deux groupes par tirage au sort : le groupe expérimental et le groupe témoin. Pour le premier, un régime pauvre en graisses saturées (d’origine animale) et riche en graisses végétales non saturées (maïs, soja…) était préconisé, avec un effet très marqué de diminution de cholestérol (15–20 %). Or, dans ces trois études, les résultats n’ont pas été à la hauteur des espérances. Dans celle d’Oslo les nombres de décès dans les deux groupes ont été de 101 et 108. Dans l’étude de Londres, la mortalité cardiaque était identique (3.5 et 3.2 décès par an). Dans l’étude de Sydney, la mortalité était plus forte dans le groupe expérimental (3.3 par an) que dans le groupe témoin (2.4), suggérant que le régime anticholestérol faisait diminuer l’espérance de vie. Il est significatif que ces études aient été rarement citées !
Dans l’étude Los Angeles Diet Trial (Dayton S. et al., 1969A11), des vétérans de la Seconde Guerre mondiale ont été répartis en un groupe expérimental (424) et un groupe témoin (422) dont les caractéristiques initiales étaient identiques selon de nombreux critères. Le groupe expérimental a été soumis pendant 8 ans à un régime pauvre en graisses saturées (38.9 % des calories en matières grasses, 365 mg par jour de cholestérol), résultant en une baisse moyenne de 12.7 % du cholestérol sanguin et une forte augmentation des acides linoléïque (oméga 6N38) et alpha-linoléique (oméga 3N39). Le groupe témoin a continué à consommer le régime standard américain (40.1 % des calories en matières grasses, 653 mg par jour de cholestérol).
Dans le groupe expérimental, les morts subites, les infarctus du myocarde et les AVC ont diminué significativement, mais la mortalité totale est restée identique (121 et 126 décès). Par contre, on a constaté plus de cancers dans le groupe expérimental (57 contre 35), ainsi que plus d’augmentation du poids moyen. D’autre part, l’incidence sur les accidents cardiovasculaires était limitée aux sujets âgés de moins de 65 ans au début de l’étude. Suivre un régime alimentaire visant à diminuer le cholestérol résulterait donc — chez de jeunes personnes — en une diminution des problèmes vasculaires, avec une augmentation des cancers et de l’obésité, sans effet notable sur l’espérance de vie. Ce détail, enfin, est important (Dayton, S. et al., 1969A11) : L’analyse de l’étendue d’athérosclérose des patients décédés n’a pas révélé de différence significative entre les deux groupes. Même remarque pour les taux de lipides et de calcium [dans les plaques d’athérosclérose].
Plus récemment, une étude observationnelle (Jakobsen MU et al., 2009A22) couvrant 344 000 personnes pendant six ans suggère que le remplacement de graisses saturées par des glucides ou des graisses mono-insaturées (huiles végétales) augmenterait le risque de maladie coronarienne. Ce résultat est corroboré par la résurgence d’un essai randomisé contrôléN40 couvrant 9423 participants, le Minnesota Coronary Experiment (1968–73) fossilisée dans les archives depuis plus de 40 ans ! Christopher E Ramsden et al. (2016A41) l’ont résumée ainsi :
Le régime à base d’huile végétale a fait baisser le taux de cholestérol, mais pas la mortalité ni les maladies cardiaques. En fait, pour les participants de plus de 65 ans, des taux de cholestérol plus faibles ont conduit à des taux de mortalité plus élevés, et non plus bas.
[Voir un article en français (Bailey R, 2016C1)].
Un autre essai clinique randomisé contrôlé sur 418 hommes, Sidney Diet Heart Study en 1966–1973, pour le remplacement des graisses saturées par des graisses mono-insaturées LA (acide linoléique oméga 6N38), avait donné le même résultat : augmentation de la mortalité cardiovasculaire 70 %, cardiaque 74 %, et 62 % pour toutes causes (Ramsden CE et al., 2013A40). Curieusement, ce résultat a aussi été « oublié » pendant 40 ans, comme tous les travaux en désaccord avec le dogme proclamé par Ancel Keys…
Plus récemment, la méta-analyse de Robert DuBroff (2018A15) portant sur 29 essais cliniques de réduction du cholestérol par les statines (postérieurs à 2004) révèle que seuls 2 essais ont affiché une (très faible) diminution de la mortalité, et un tiers une (infime) réduction des maladies cardiovasculaires. L’auteur conclut (p. 996) :
Trois décennies d’essais contrôlés randomisés ont […] donné des résultats incohérents et contradictoires. Nous devons reconnaître ces anomalies et modifier ou rejeter l’hypothèse lipidique. Il est clair que certaines personnes bénéficient d’un traitement modifiant les lipides. Je pense que la vraie question est de savoir comment les identifier. Notre approche actuelle, qui consiste à se concentrer presque exclusivement sur la réduction du LDL‑C pour tout le monde, n’est pas toujours efficace, peut entraîner le traitement inutile de certaines personnes en bonne santé, et reflète probablement le fait que la pathogenèse de l’athérosclérose est beaucoup plus complexe qu’on ne le pensait à l’origine. Notre approche de la prévention des maladies cardiovasculaires centrée sur le LDL‑C nous a peut-être détournés de l’étude d’autres mécanismes physiopathologiques et d’autres traitements.
Une discussion plus détaillée des essais cliniques se trouve dans mon article Statines et médicaments anticholestérol. Un excellent résumé a été réalisé, dans une vidéo de 24 minutes, par le Dr Pierre-Vladimir Ennezat (2018C5). Une autre vidéo (2025N41) couvre le sujet plus vaste de la santé métabolique.
⇪ Mourir jeune et en bonne santé ?
Plusieurs études ont mesuré que la longévité de personnes âgées était supérieure chez celles qui ont un taux élevé de LDL‑C. Dans cette population, le cholestérol — global aussi bien que LDL‑C — serait protecteur contre les infections et l’athérosclérose (Ravnskov U, 2003A42 ; Ravnskov U et al., 2016A43) :
Un LDL‑C élevé est inversement associé à la mortalité chez la plupart des personnes de plus de 60 ans. Cette constatation est incompatible avec l’hypothèse du cholestérol (c.-à‑d. que le cholestérol, en particulier le LDL‑C, est intrinsèquement athérogène). Étant donné que les personnes âgées avec un LDL‑C élevé vivent aussi longtemps ou plus longtemps que celles avec un LDL‑C faible, notre analyse fournit des raisons de remettre en question la validité de l’hypothèse du cholestérol.
Il est utile de rappeler que les études cliniques sur les médicaments anticholestérol ne sont conduites que sur une population de 59 ± 10 ans en moyenne. En 2018, l’équipe d’Alessandra Errigo (2025N42) a recruté 168 nonagénaires de référence (81 hommes, 87 femmes) dans une zone bleue de longévité située en Sardaigne, qu’elle a suivis jusqu’en décembre 2024. Résultat :
La médiane du cholestérol total était de 199.5 (intervalle 89–314) mg/dL chez les hommes et de 202.5 (intervalle 89–324) mg/dL chez les femmes. La durée de survie était significativement plus longue chez les participants dont le cholestérol LDL (LDL‑C) était supérieur à 130 mg/dL que chez les nonagénaires dont le LDL‑C était inférieur à 130 mg/dL (3.82 ± 1,88 ans contre 2.79 ± 1,56 ans, p < 0.0001). L’analyse de régression de Cox a révélé une réduction significative du ratio de risque (HR) de mortalité chez les participants atteints d’hypercholestérolémie légère (LDL‑C ≥ 130 mg/dL) par rapport à celui des participants ayant un taux de cholestérol normal (OR 0.600, 95 % IC 0.405–0.891).
Conclusions : Dans la population à longue durée de vie examinée, il est peu probable que le paradoxe du cholestérol soit le reflet d’une causalité inverse. Nos résultats remettent en question l’opinion courante selon laquelle la longévité est invariablement associée à un faible taux de cholestérol. En outre, une hypercholestérolémie modérée n’empêche pas l’adulte le plus âgé d’atteindre un âge avancé, contrairement à ce que l’on croit souvent.
À l’inverse de ces résultats, il est utile de citer l’article de Dong D. Wang et collègues (2016A56) du département de nutrition de Harvard T. H. Chan School of Public Health dirigé par Frank HuN43. En s’appuyant sur les données du Nurses’ Health Study, cet article concluait à un avantage, en termes de mortalité toutes causes, de remplacer les graisses saturées (et trans) par des huiles insaturées (végétales). Or il a été épinglé sur PubPeer comme entaché d’erreursN44. Ceci, à la fois pour le traitement des données (conclusions en contradiction avec les données) et parce qu’il repose entièrement sur des collectes de données « basées sur la mémoire » — voir mon article Faut-il jeter les enquêtes nutritionnelles ? Edward Archer souligne (2016N44) : Par exemple, les régimes apparaissant dans des études épidémiologiques comme Nurses’ Health Study ne permettraient pas la survie s’ils étaient consommés quotidiennement. Un autre commentateur signale les liens d’intérêt entre Frank Hu et la California Walnut CommissionN45 qui fait la promotion des producteurs de noix en Californie.
Une étude rétrospective récente (Potluri R et al., 2017A38) basée sur le suivi pendant 14 ans d’un million de femmes d’âge moyen 66 ans au nord-ouest de l’Angleterre, a comparé un groupe de patientes avec cholestérol élevé avec celui à faible cholestérol. L’étude statistique a montré que, dans le groupe à haut cholestérol, l’incidence de cancer du sein avait été 0.6 fois celle de celui à faible cholestérol. Le groupe diagnostiqué à haut cholestérol au départ avait aussi une mortalité plus faible que celui à bas cholestérol (13.8 % contre 23.7 %) (Paddock C, 2017A36).
Selon Rahul Potluri et collègues (2017A38) :
Un modèle de régression logistique tenant compte du temps entre la première présentation et le développement du cancer du sein a montré que la présence d’hyperlipidémie améliorait le résultat du cancer du sein de 1.64 fois (95 % C.I. 1.50–1.79).
Potluri et collègues ont visiblement été perturbés par ce résultat qui va à l’encontre de ce qu’ils espéraient… Ils n’hésitent donc pas avancer une hypothèse hasardeuse :
Si un diagnostic de taux de cholestérol élevé entraîne une baisse des taux de cancer du sein, cela doit être relié à quelque chose d’inhérent dans la condition des patientes affectées, ou plus probablement, à un traitement par des médicaments largement utilisés pour diminuer le cholestérol, comme les statines.
Une étude suédoise (Heir T et al., 2016A20) correspondant au suivi de 2000 hommes sur 40 années met aussi en évidence une association inverse entre le niveau de cholestérol total et le risque de cancer de la prostate :
Pour le cancer de la prostate au stade avancé et général, l’incidence était deux fois plus élevée dans le quartile inférieur de cholestérol [1.04 – 2.23 g/l] comparé au quartile le plus élevé [2.89 – 5.94 g/l]. Ces associations sont demeurées significatives après ajustement pour l’âge, le tabagisme, la condition physique, l’IMC et la pression artérielle systolique.
La revue systématique de Chowdhury R et al. (2014A9) sur les associations du risque coronaire avec la consommation de graisses saturées conclut :
Les preuves actuelles ne soutiennent pas clairement les directives de médecine cardiovasculaire encourageant une consommation élevée d’acides gras polyinsaturés et une faible consommation de graisses saturées totales.
Alan Rozanski et ses collègues (2022A47) ont observé la relation entre la mortalité et l’hypercholestérolémie ou d’autres facteurs de risque de maladies coronariennes, sur un large éventail de patients orientés vers divers examens cardiaques. Le titre de leur article est : Association entre l’hypercholestérolémie et le risque de mortalité chez les patients orientés vers un examen d’imagerie cardiaque : Preuve d’un « paradoxe du cholestérol » ?
Cette étude couvrait 64 357 patients soumis à une scintigraphie des artères coronaires (CAC), 10 814 patients soumis à une angiographie coronarienne par tomodensitométrie (CCTA), 31 411 patients sans maladie coronarienne connue soumis à une imagerie de perfusion myocardique (MPI) par tomographie d’émission monophotonique (SPECT) à l’effort/au repos, et 5051 patients avec une maladie coronarienne connue soumis à une SPECT-MPI à l’effort/au repos. Chacune de ces cohortes a été suivie pour la mortalité toutes causes confondues en utilisant des modèles de Cox ajustés au risqueN46.
L’utilisation du terme « paradoxe » est emblématique d’un résultat qui ne fait rien d’autre que contredire une croyance dominante. Ils écrivent (Rozanski A et al., 2022A47 p. 61) :
Dans des données d’observation concernant des patients orientés vers une épreuve d’effort cardiaque, on a noté une relation inverse entre des antécédents d’hypercholestérolémie et la mortalité ultérieure […]. En outre, dans certaines cohortes communautaires récentes, une relation non linéaire a été observée entre les taux sériques de cholestérol à lipoprotéines de basse densité (LDL‑C) et la mortalité.
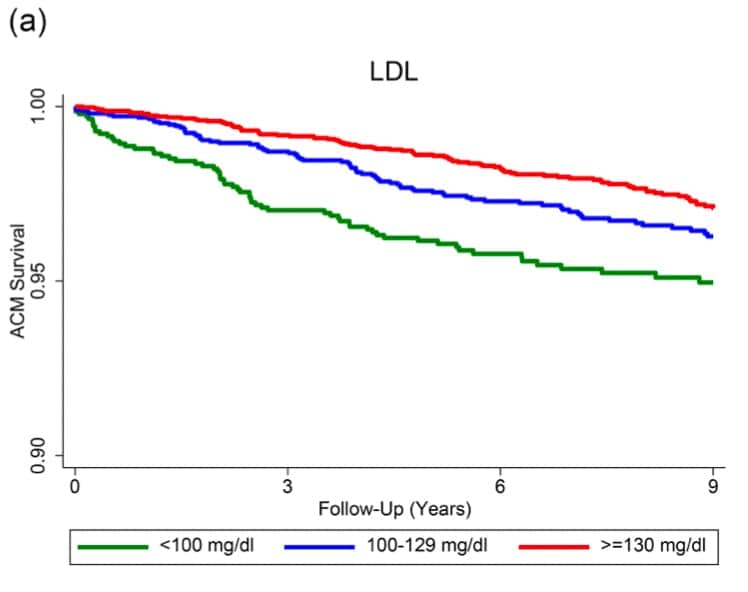
ACM = all-cause mortality
L’image ci-contre (Rozanski A et al., 2022A47 p. 67) montre la décroissance des populations sur 9 années d’observation en distinguant celles à haut (≥ 130 mg/dl), moyen (100–129 mg/dl) et bas (< 100 mg/dl) taux de LDL‑C. Il est clair que les patients à haut taux de cholestérol LDL‑C ont été plus nombreux à résister aux décès pour toutes causes…
Dans les tableaux à la fin de l’étude qui comparent un facteur de risque à un autre, on peut constater qu’un taux élevé de cholestérol LDL réduit en fait les risques de mourir plus tôt du diabète, et qu’il égalise les risques de mourir plus tôt en tant que fumeur. Toutefois, les chercheurs ont envisagé une autre hypothèse qui résoudrait le « paradoxe » :
Compte tenu de l’omniprésence et de l’efficacité des traitements hypolipidémiants, il est possible que nos résultats soient dus à ce que l’on pourrait appeler un « biais de traitement ». Ce biais est introduit lorsqu’un prédicteur de risque efficace entraîne des interventions thérapeutiques puissantes qui modifient ensuite les résultats utilisés pour évaluer l’importance clinique du prédicteur de risque mesuré au cours d’une étude ultérieure. Comme nous n’avons pas eu accès aux médicaments initiés après la réalisation de nos tests non invasifs, nous n’avons pas pu rendre compte de l’ampleur de ce biais dans notre étude, mais il est probable qu’il ait été puissant. Cependant, il est peu probable qu’un tel biais de traitement explique entièrement la relation inverse que nous avons observée entre les taux de cholestérol et la mortalité, en particulier à la lumière de la robustesse de nos résultats dans un large éventail de cohortes qui variaient considérablement en termes de risque clinique.
Cette hypothèse explicative du « paradoxe » a été — hélas pour eux — contredite par les données :
[…] lorsque nous avons limité nos résultats aux patients étudiés entre 1991 et 1995, une période précédant l’introduction des statines les plus puissantes d’aujourd’hui, comme l’atorvastatine et la rosuvastatine [deux statines précoces], nous avons constaté des résultats similaires.
⇪ Hypercholestérolémie familiale
L’hypercholestérolémie familialeN47 — une caractéristique génétique — est souvent citée comme une cause de maladie cardiovasculaire en raison de l’excès de cholestérol qui la caractérise. Il est vrai que, selon la méta-analyse de Pengwei Hu et al. (2020A21), parmi les personnes souffrant d’athérosclérose figurent plus fréquemment celles qui présentent une hypercholestérolémie familiale (HF). Mais cette fragilité est-elle un effet du cholestérol ?
Michel de Lorgeril (2024C2) montre que ce lien n’est pas confirmé par les données, en s’appuyant sur la publication de Dennis Kumi et collègues (2024A29): parmi 3.7 millions d’infarctus, les auteurs identifient 2360 HF donc 0,06 % du total, [alors que] l’incidence de l’HF dans nos populations est d’environ 0,32 %. Il y a donc, chez les personnes atteintes d’hypercholestérolémie familiale, cinq fois moins d’infarctus que ce qui était prévisible. De Lorgeril ajoute (2024C2) :
1) les HF font globalement moins d’infarctus mais font proportionnellement plus (60 % d’augmentation du risque relatif) d’infarctus de type 1 (par définition, obstruction coronaire totale par un thrombus) par rapport aux non-HF ;
2) les HF font aussi proportionnellement moins (39 % de réduction) d’infarctus de type 2 (pas d’obstruction coronaire ; pas de thrombose identifiable) par rapport aux non-HF ;
3) les HF font enfin plus (3 fois plus) de thrombose intracardiaque par rapport aux non-HF quand ils font un infarctus.
Dit autrement, les HF font beaucoup moins d’infarctus (que ce qui est attendu) et quand ils en font, ils sont visiblement plus liés à des troubles de type hypercoagulabilité.
Finalement, et c’est le point le plus important, la mortalité des HF dans l’étude de Kumi est réduite de moitié par rapport aux non-HF.
[…] ces données confirment ce que nous savions : les infarctus dans l’HF ne sont pas dépendants du niveau de cholestérol mais d’une prédisposition à l’hypercoagulation dont les causes peuvent être génétiques (à rechercher et neutraliser) ou conséquences d’un mode de vie délétère qu’il faut modifier.
Les personnes qui présentent une hypercholestérolémie familiale ont aussi une lipoprotéine(a) élevée qui augmente le risque de caillot artériel (De Lorgeril M, 2011B1 p. 116 ; 2022B4 p. 42–43). L’hypercoagulation est souvent aggravée par le manque d’exercice — voir Soigner ses artères.
⇪ Données mondiales
On peut apprécier le travail du blogueur Ricardo Carvalho pour son interprétation graphique des données de statistiques de mortalité de l’OMS en 2002N48 associées, dans 164 pays, aux taux de cholestérol fournis en 2005 par la British Heart Foundation, ce qui aboutit sans surprise à des courbes en UN49 :
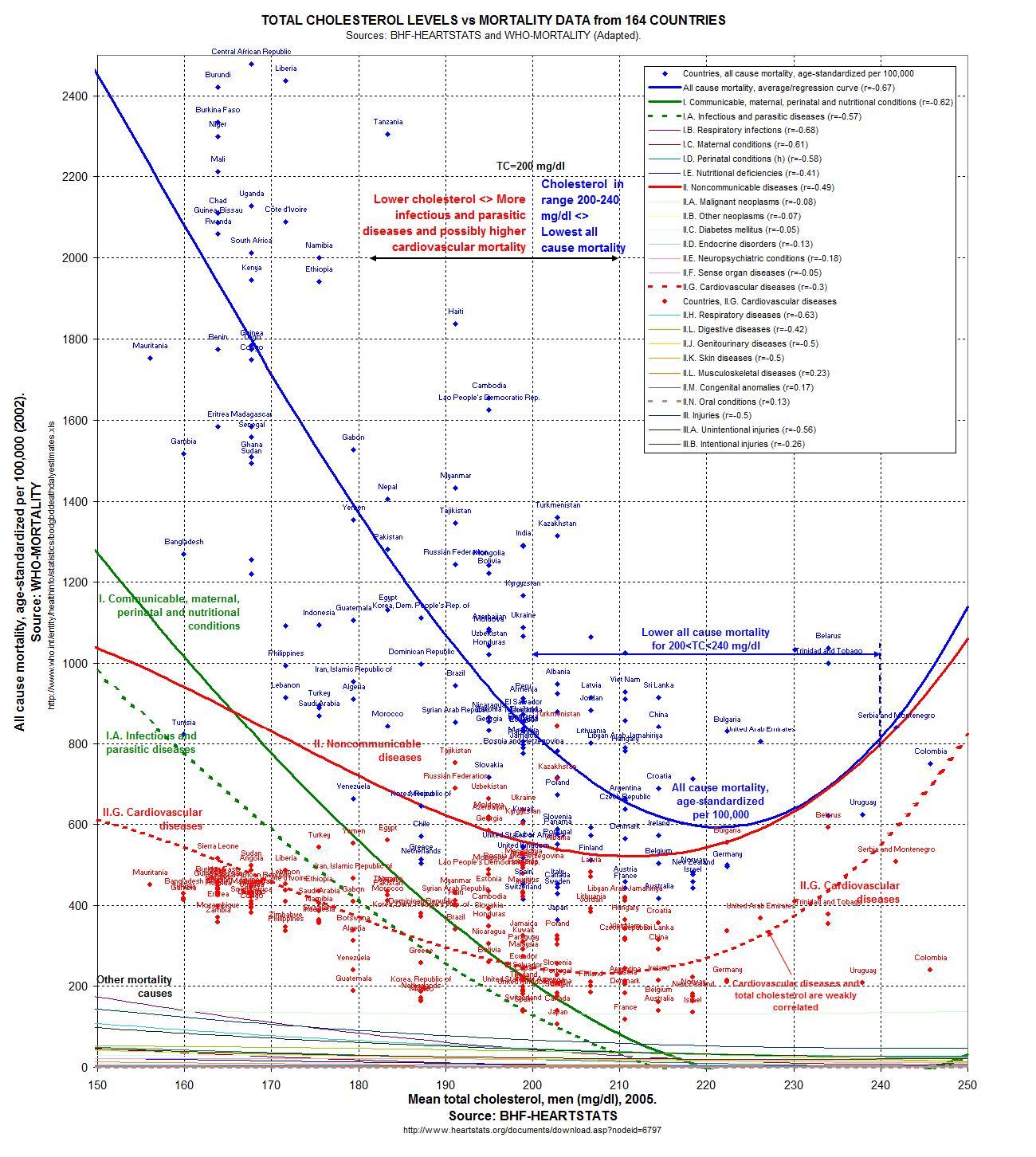
Le graphique montre que la valeur optimale du taux de cholestérol pour minimiser la mortalité toutes causes confondues serait dans la fourchette 200–240 mg/dl alors qu’en France, par exemple, il est recommandé de le maintenir à moins de 200 mg/dl.
Il est intéressant de noter que la France et le Japon affichent les meilleurs résultats en termes de mortalité par maladie cardiovasculaire (courbe pointillée en rouge) et toutes causes confondues (courbe solide en bleu) avec des taux de cholestérol respectifs de 210 et 202 mg/dl, autrement dit au-dessus du « seuil de dangerosité » en France.
La ligne en vert pointillé montre que le taux de cholestérol est inversement corrélé avec la mortalité par maladie infectieuse ou parasitaire — ce qui est logique puisqu’il est un des ingrédients du système immunitaire, voir mon article Soigner ses artères.
Un graphique similaire présente les taux de décès pour toutes causes en fonction des taux sanguins de cholestérol total pour des personnes de 15 à 60 ans dans 181 pays :
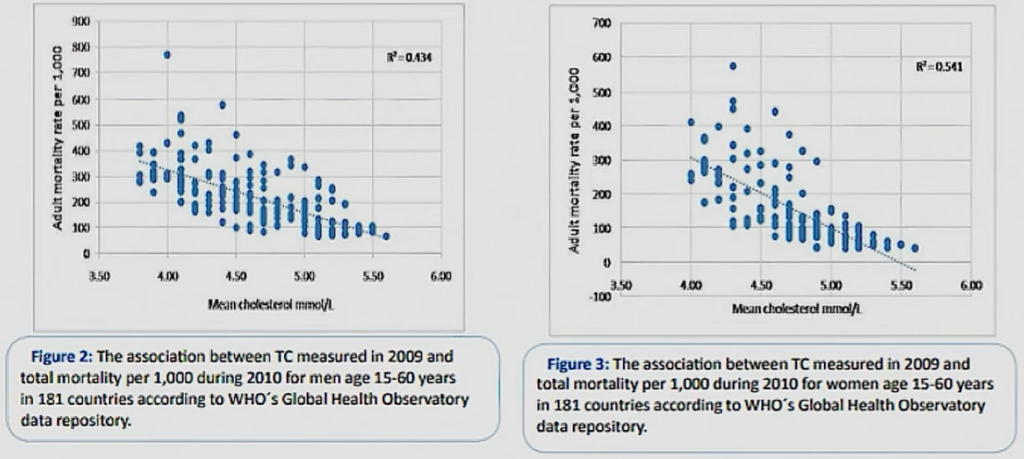
Le taux de 5.00 mmol/l correspond à 193 mg/dl.
Ces diagrammes sont conformes aux données affichées ci-dessus par Ricardo Carvalho, qui les avait résumées par une régression quadratique (parabole en bleu) au lieu d’une régression linéaire.
Les données affichées par Xuan-Mai T Nguyen et al. (2023A35) sont peu concluantes — bien qu’elles portent sur 4 millions de vétérans de l’armée américaine — car ces auteurs ne se sont intéressés qu’à la mortalité cardiovasculaire, et non la mortalité pour toutes causes qui est bien plus significative.
Un tableau (de source inconnue) présenté dans une intervention de Malcolm Kendrick (2023C6) démolit la croyance selon laquelle « manger gras serait mauvais pour le cœur ». Il compare la mortalité cardiovasculaire dans plusieurs pays au pourcentage calorique de la consommation de matières grasses :
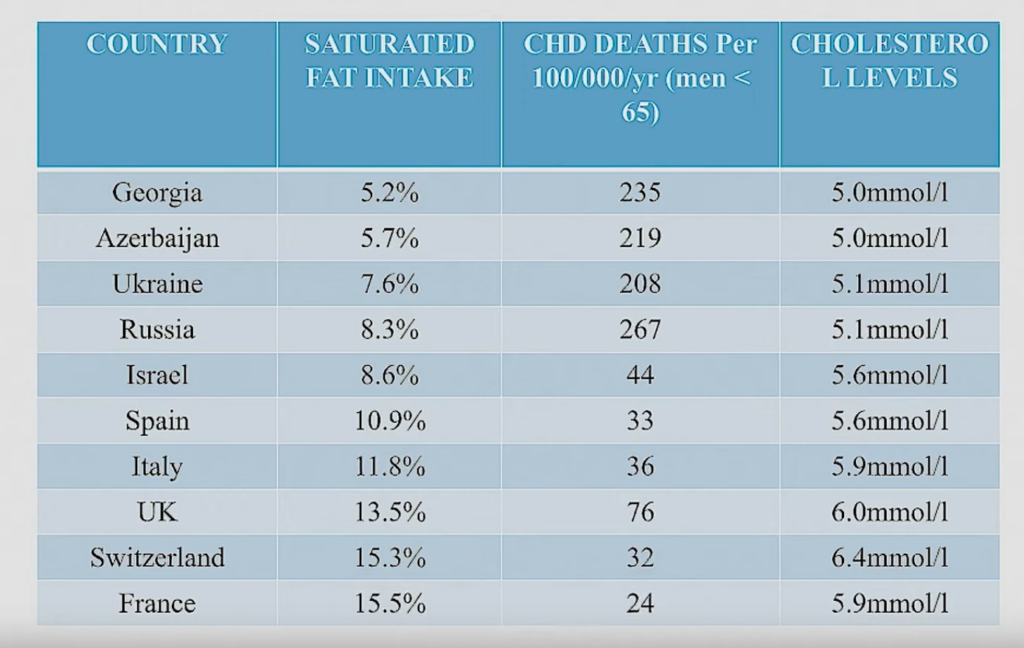
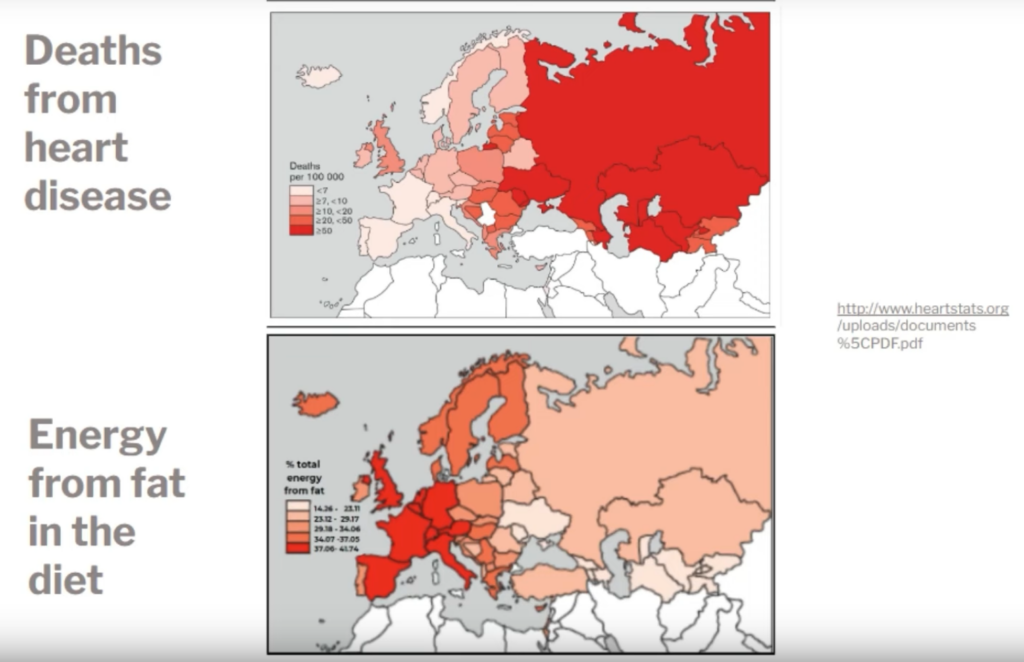
Dans le même exposé, Kendrick (2023C6) a présenté deux cartes d’Europe illustrant les taux de mortalité cardiovasculaire et les pourcentages d’énergie puisée dans les matières grasses :
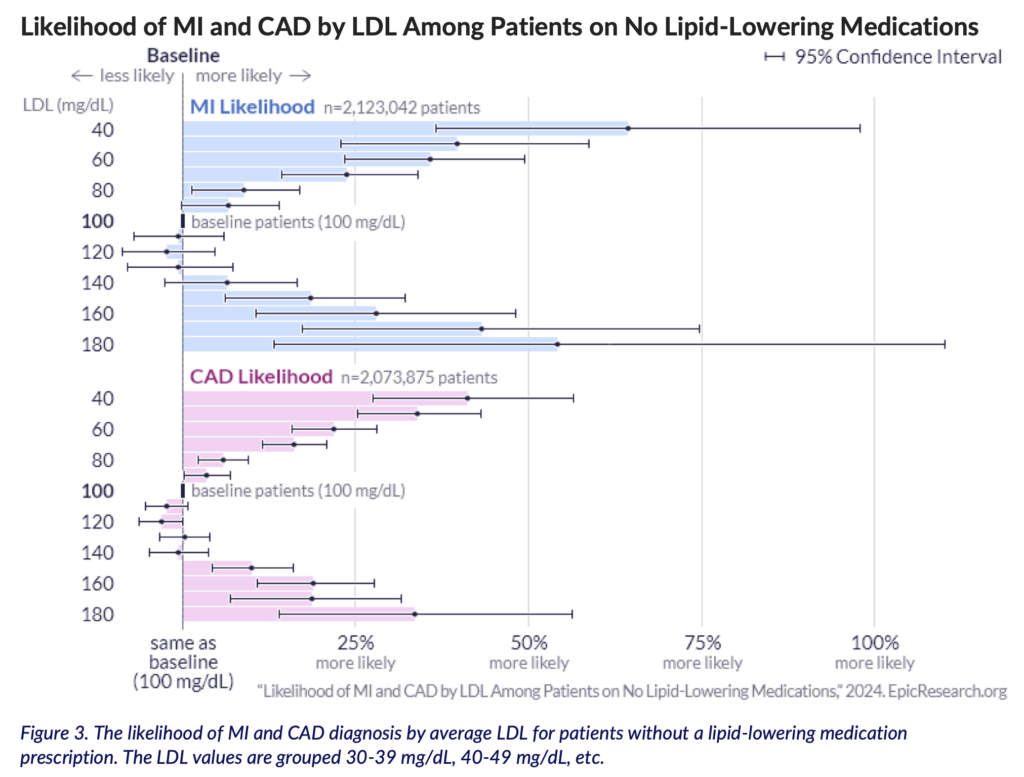
Une étude de plus de 256 millions de résultats d’analyse dans 50 États des USA et au Liban montre que, pour des patients non traités pour dyslipidémie, la fourchette optimale du taux de LDL pour minimiser les risques d’infarctus (MI) et de maladie cardiovasculaire (CAD) se situerait aux alentours de 100 à 140 mg/dl (Bartelt K et al., 2024N50) :
Une liste classifiée de publications démontrant l’inutilité de « faire baisser le cholestérol » a été publiée par Pascal Raton (2017C7).
⇪ Longévité des croyances
Malgré tous ces résultats, des études comme celle de Johansson, I et al. (2012A23) portant sur 140 000 observations en 25 ans de la population suédoise, affirment (sur la base de seules corrélations issues de questionnaires nutritionnels) que l’augmentation des taux de cholestérol dans la période 2004–2009 — attribuée à la popularisation des régimes low carb/high fat — serait un facteur de risque cardiovasculaire. Mais ils en tirent un raisonnement d’une naïveté déconcertante :
[…] la baisse prometteuse de la mortalité par maladie cardiovasculaire au cours des 20 dernières années a été attribuée principalement à des diminutions salutaires du cholestérol sanguin, des triglycérides, du tabagisme et de l’hypertension. La diminution du cholestérol seul explique 39 % de la réduction de la mortalité. Ainsi, la tendance à la hausse observée depuis 2004 et l’augmentation marquée du cholestérol après 2007 sont une source de profonde préoccupation pour la prévention primaire et secondaire des maladies coronariennes.
On peut écouter le début d’un entretien de Joe Rogan avec le cardiologue Dr Aseem Malhotra, l’un des premiers médecins, au Royaume-Uni, à critiquer publiquement le « mythe du cholestérol » et la prescription de statines (Rogan J, 2023N51).
Les anglophones apprécieront aussi l’intervention (désopilante) du Dr Malcolm Kendrick intitulée « Perte de foi en la recherche médicale » (2023C6). Car ce n’est pas qu’une question d’argent — les milliards de dollars engrangés par l’industrie pharmaceutique dans la vente de statines — mais aussi de religion. Kendrick écrit (2021B6 p. 34) :
L’Église adventiste du septième jour est depuis longtemps un acteur important de la recherche nutritionnelle.
Il cite ensuite Jim E Banta et al. , auteurs adventistes (2018A4) :
L’accent mis sur le ministère de la santé au sein du mouvement adventiste du septième jour (SDA) a conduit au développement de sanatoriums dans l’Amérique du milieu du dix-neuvième siècle. Ces établissements, dont le plus remarquable est celui de Battle Creek, dans le Michigan, ont été à l’origine du développement d’aliments végétariens, tels que les céréales pour le petit-déjeuner et les imitations de viandes. L’Église SDA exploite encore une poignée d’installations de production alimentaire dans le monde. Le premier diététicien du Battle Creek Sanitarium a été cofondateur de l’American Dietetics Association, qui a fini par préconiser un régime végétarien.
L’Église SDA a créé des centaines d’hôpitaux, de collèges et d’écoles secondaires et des dizaines de milliers d’églises dans le monde entier, qui prônent tous un régime végétarien. Dans le cadre du « message de santé », le régime alimentaire continue d’être un aspect important des efforts d’évangélisation de l’Église. Outre la promotion d’un régime végétarien et de l’abstinence d’alcool, l’Église adventiste a également investi des ressources pour démontrer les avantages de ces pratiques pour la santé par le biais de la recherche. La plupart de ces recherches ont été menées à l’université de Loma Linda, dans le sud de la Californie, où trois études de cohortes prospectives ont été réalisées sur une période de 50 ans.
Malcolm Kendrick commente (2021B6 p. 35) :
Les adventistes nous ont également donné cet objet croustillant et insipide connu sous le nom de cornflake. […]
L’argent et la religion, voilà ce que j’appelle une puissante combinaison. Ensuite, lorsque les statines sont apparues, avec leur capacité unique à réduire de manière significative le taux de cholestérol, toute la puissance de l’industrie pharmaceutique a mis les bouchées doubles. Il ne s’agit pas d’une simple industrie d’un milliard de dollars, mais d’une industrie d’un billion de dollars. Il y a aussi l’industrie des aliments pauvres en graisses, qui pèse des milliers de milliards de dollars.
Donc, non, il n’y a pas de grande cabale mondiale, maléfique et obscure, promouvant l’hypothèse du cholestérol. Mais il y a des acteurs de taille, qui se rangent presque tous du même côté de l’argument. Beaucoup d’entre eux sont bien intentionnés, d’autres le sont nettement moins.
La science s’oppose à tout cela. Du moins, je crois que c’est la science. Du côté de la science, il y a ces choses terribles et gênantes que l’on appelle « les faits ».
L’influence de l’Église adventiste du septième jour sur la recherche nutritionnelle nord-américaine est décrite plus en détail dans mes articles Hunza à perte de vue et Pour les végan·e·s.
⇪ Vers un abandon du modèle « plomberie » ?
J’ai entendu de la bouche d’un « expert YouTube » le récit le plus désopilant — ou désolant ? — sur la formation de la plaque d’athérosclérose. Ce médecin (!) disait que le sucre consommé dans nos aliments sédimente en de minuscules cristaux à la surface des vaisseaux sanguins, cristaux qui ont la fâcheuse propriété d’accrocher « le cholestérol en circulation dans le sang »… (Fin de plaisanterie)
Selon Michel de Lorgeril (page Wikipedia avant 2017N52) :
La réalité médicale montre que la phrase « le cholestérol bouche les artères » est un résumé, grossier et faux, de la complexité physiologique. Une plaque d’athérosclérose n’est pas constituée uniquement de cholestérol. Celui-ci occupe un volume entre allant 0 % à 20 % de la plaque. Le cholestérol, présent dans le sang au sein des transporteurs (VLDLN24, LDLN22, Lp(a)N25, HDLN21…), sera aggloméré dans la plaque d’athérosclérose si le transporteur est abîmé (oxydé et/ou glycosyléN53). Mais les accidents vasculaires semblent plutôt liés à une inflammation répétée de l’épithéliumN54 des vaisseaux sanguins, inflammation que l’alimentation, les glucides, l’obésité, le stress et la pollution induisent fortement.
Sylvain Duval commentait (2013A16) :
Cela signifie que le régime alimentaire peut être une bonne mesure hygiéno-diététique, si la cible n’est pas de viser une baisse du cholestérol. La clef de la réussite des régimes se trouve ailleurs que dans la théorie du cholestérol.
La formation de plaque d’athérosclérose a été souvent associée à l’inflammation systémiqueN55 — voir à ce sujet l’étude historique de Sergio Minelli et collègues (2020A31) :
Le dysfonctionnement endothélial, qui est à la base de la maladie, est prédisposé par de nombreux facteurs de risque (tels que le LDL modifié ou oxydé, le diabète sucré, l’hypertension, le tabagisme, les agents infectieux, l’âge et l’association de ces facteurs ou d’autres) et implique des troubles des propriétés antithrombotiques, pro-fibrinolytiques, anti-inflammatoires et anti-oxydantes de l’endothélium. […]
La réponse inflammatoire et à médiation immunitaire peut agir soit en favorisant l’athérosclérose, soit en facilitant la guérison des lésions ; de plus, elle influence la coagulation et la fibrinolyse, pouvant ainsi moduler les complications thrombotiques de la maladie. […]
De nombreux médicaments anti-inflammatoires, tels que les statines, les antithrombotiques et les antihypertenseurs sont largement utilisés, mais au mieux, ils ne font que retarder la progression de l’athérosclérose ; par conséquent, de nouvelles thérapies anti-athérosclérotiques qui s’attaquent au risque inflammatoire résiduel sont justifiées.
Michael B. Rothberg (2013A46) prône l’abandon du modèle simpliste décrivant la maladie cardiovasculaire comme le simple effet d’un rétrécissement des artères : un « problème de plomberie » qui serait résolu par une revascularisation N56 :
Nous savons que les interactions entre la graisse diététique, le cholestérol sérique et l’endothélium artérielN57 sont complexes et dynamiques. Bien que les sténosesN58 de haut niveau puissent provoquer une angineN59 chronique, la plupart des événements cardiaques se produisent lors de lésions apparues légères lors de l’angiographieN12 précédente. Ces plaques contiennent un noyau riche en lipides recouvert d’un bouchon fibromique mince. Les cellules inflammatoires (par ex. les macrophagesN60 et les mastocytesN61) dans la plaque peuvent être activées par des microbes, des autoantigènesN62 ou des molécules inflammatoires (modèle de la plaque activée). Les cellules activées sécrètent des cytokinesN63 et des protéasesN64 qui affaiblissent le capuchon fibreux, ce qui l’entraîne à s’éroder ou à se rompre. Le sous-endothéliumN57 nouvellement exposé et les facteurs procoagulants précipitent l’agrégation plaquettaire et la formation locale de thrombusN16, entraînant parfois un infarctus. Avant la rupture, ces plaques ne limitent pas souvent l’écoulement et peuvent être invisibles à l’angiographieN12 et aux tests de stressN14. Elles ne sont donc pas susceptibles d’intervention coronarienne percutanée.
Chriss Kresser (2022A28) suggère qu’un meilleur prédicteur de maladie cardiovasculaire ne serait pas le taux de cholestérol dans les lipoprotéines de faible densité (en abrégé, LDL‑C) mais plutôt le nombre de particules LDL (LDL‑P) :
Les lipoprotéinesN20 sont comme des automobiles qui portent le cholestérol et les graisses autour de votre corps, et le cholestérol et les graisses sont comme des passagers dans ces automobiles. Les scientifiques ont l’habitude de croire que le nombre de passagers dans l’automobile (c’est-à-dire la concentration de cholestérol dans la particule de LDL) est le facteur déterminant dans le développement de maladies cardiaques. Des études plus récentes, cependant, suggèrent que c’est le nombre d’automobiles sur la route (c’est-à-dire les particules de LDL) qui comptent le plus.
Les artères coronaires sont essentiellement des tubes creux, et l’endothélium (doublure) de l’artère est très mince, l’épaisseur d’une seule cellule. Le sang, qui transporte des lipoprotéines comme le LDL, est en contact constant avec la doublure endothéliale. Alors, pourquoi la particule LDL quitte-t-elle le sang pour pénétrer dans l’endothéliumN57 et entrer dans la paroi de l’artère ? La réponse est que c’est un phénomène de gradient. En revenant à notre analogie, plus il y a d’automobiles sur la route, plus il est probable que certaines d’entre elles vont « s’écraser » dans la fragilité de l’artère. Ce n’est pas le nombre de passagers (cholestérol) que les automobiles transportent qui est le facteur déterminant, mais le nombre d’automobiles sur l’autoroute.
Michael H. Davidson et collègues (2011A10) ont vérifié que les patients ayant un cholestérol LDL élevé (LDL‑C) et un faible nombre de particules LDL (LDL‑P) ne présentaient pas un risque élevé de maladie cardiaque. Au contraire, ils courent un moindre risque que les patients à faible taux de LDL‑C et à taux élevé de LDL‑P.
Cette remise en cause du LDL‑C comme prédicteur d’accidents cardiovasculaires est clairement exposée sur la fiche LDL de Wikipedia en anglaisN65 — alors que la fiche francophone l’ignore entièrement :
La concentration des particules de LDL [LDL‑P] et, dans une moindre mesure, leur taille, a une corrélation plus forte et cohérente avec le résultat clinique individuel que la quantité de cholestérol dans les particules de LDL, même si l’estimation du LDL‑C est approximativement correcte.
Ces résultats sont exposés et commentés — sur un ton humoristique — par David M. Diamond sur sa vidéo An Update on Demonization and Deception in Research on Saturated Fat (2017N66 7:08).
Kresser identifie par ailleurs cinq causes d’un taux élevé de LDL‑P : l’insulinorésistanceN67, le syndrome métaboliqueN68, un mauvais fonctionnement de la thyroïde, des infections bactériennes ou virales, le syndrome de porosité du colonN69 ou une anomalie génétique.
Enfin, le taux de LDL oxydé (oxLDL) serait un prédicteur d’athérosclérose chez les personnes âgées, voir Dayuan Li & Jawahar L. Mehta (2005A30).
⇪ Composition des plaques
L’image populaire de l’athérosclérose — entretenue par les marchands de statines — est celle de plaques formées par l’accumulation de « cholestérol » qui progressivement « bouche les artères »… Il est prudent de confronter cette croyance aux faits.
David Gertz et ses collègues ont analysé pour cela les cœurs et les artères coronaires de 18 patients décédés à plus de 90 ans (1991A18 p. 1228) :
Les patients de cette étude avaient été inclus dans une précédente étude de ce laboratoire portant sur les caractéristiques cliniques et morphologiques cardiaques de 40 patients ≥ 90 ans […]. Les patients ayant subi un pontage coronarien ou une angioplastie coronarienne transluminale percutanée ont été exclus. Onze patients présentaient des infarctus du myocarde (aigus, cicatrisés ou les deux) à l’autopsie, et 7 patients décédés de causes non cardiaques ne présentaient pas d’infarctus à l’autopsie.
Les plaques ainsi analysées était donc celles de la « phase terminale » de la maladie cardiovasculaire qui avait entraîné le décès de la plupart des patients. Leur composition était la suivante :
- tissus fibreux : 87 ± 8 %
- dépôts calcaires : 7 ± 6 %
- débris pultacés : 5 ± 4 %
- cellules spumeusesN70 : 1 ± 1 %
Les débris spongieux étaient des « zones de coloration pâle avec d’abondantes stries de cholestérol, contenant ou non des érythrocytesN71 [globules rouges] et des cellules inflammatoires » (Gertz D et al., 1991A18 p. 1231).
Notons que, même si du cholestérol est présent, il n’apparaît que dans une proportion sans rapport avec la croyance dominante…
D’autres croyances sont à corriger, comme l’expose Malcolm Kendrick (2021B6 p. 50–53, 56–69) :
- Le LDL ne peut pas traverser l’endothélium. Pour commencer, il ne peut pas passer à travers les cellules endothéliales car, s’il parvient à y pénétrer, accroché à un récepteur de LDL, il est décomposé dans la cellule, et seul le récepteur est restitué intact (Kendrick M, 2021B6 p. 51–52). D’autre part, il ne peut pas s’insérer entre ces cellules qui sont collées l’une à l’autre par des jonctions serréesN72 (p. 53). Toutefois, il peut pénétrer la paroi d’une artère (ou d’une veine) en passant « par derrière » : les vasa vasorumN73 qui sont des capillaires irriguant les vaisseaux : des « vaisseaux de vaisseaux » (p. 88–89).
- Les stries grasses (fatty streaksN74) ne sont pas des amorces de plaque d’athérosclérose. On en trouve déjà chez tous les enfants de moins de trois ans, et même dans le fœtus, là où le taux de LDL est moins du tiers du niveau de celui de l’adulte. De plus, malgré leur appellation, elles n’ont rien de gras : ce sont des infiltrats de cellules blanches, les leucocytes (de Lorgeril M, 2011B1 p. 93). Ces stries ne se prêtent pas à une conversion directe en une lésion pouvant avoir une signification clinique (Velican C & D Velican, 1983A54). Les stries grasses et les plaques fibreuses d’athérosclérose ont des voies de développement totalement indépendantes (Kendrick M, 2021B6 p. 58).
- Les cristaux de cholestérol qui se trouvent dans les cellules spumeusesN70 (foam cells) des plaques d’athérosclérose ne proviennent pas du LDL, car pour faire des cristaux il faut du cholestérol « libre », alors que celui du LDL est toujours associé à un acide gras, formant un esterN75 de cholestérol. Le cholestérol libre est contenu, en forte quantité, dans les globules rouges (érythrocytesN71). Ces globules rouges forment l’essentiel des caillots qui en s’agglomérant contribuent à la formation des plaques — voir ci-dessous l’hypothèse thrombogénique (Kendrick M, 2021B6 p. 59–64).
- Les lipoprotéines de basse densité présentes dans les plaques d’athérosclérose ont été abusivement identifiées comme du LDL, alors que la présence d’apolipoprotéine(a) les identifie comme des lipoprotéines(a)N25 (Rath M et al., 1989N76 ; Kendrick M, 2021B6 p. 64–69). Voir ci-dessous les propriétés de ces Lp(a).
Un article détaillé : Le rôle clé de l’endothélium (de Lorgeril M, 2022B4 p. 57–72) présente les rôles protecteurs de l’endothélium — production d’oxyde nitrique (NO) et de prostacyclineN77 — ainsi que ses dysfonctionnements. Toutefois, le rôle bénéfique de l’acide linoléiqueN78 (LA) polyinsaturé y est à mon avis surestimé — voir à ce sujet l’étude de James J DiNicolantonio et James H O’Keefe (2018A14) et mon article Glucides ou lipides ?
La protection de l’endothélium est exposée dans le chapitre 10 de The Clot Thickens (Kendrick M, 2022bA27 p. 221–253).
⇪ L’hypothèse thrombogénique
Selon WikipediaN79, la thrombogénicité désigne la tendance d’un matériau en contact avec le sang à produire un thrombusN16, ou caillot. Elle ne se réfère pas seulement aux thrombus fixes mais aussi aux emboles, des thrombus qui se sont détachés et qui voyagent dans la circulation sanguine.

The Clot Thickens (le caillot s’épaissit) est un calembour évocateur de the plot thickens (l’intrigue se complique)
L’hypothèse thrombogénique sur la formation de plaque d’athérosclérose a été énoncée ainsi par Malcolm Kendrick (2022A27 p. 428) :
Un modèle alternatif [à celui du cholestérol] a été proposé pour la première fois il y a plus de cent cinquante ans par Karl von Rokitansky, à savoir que les plaques d’athérome résultent de l’accumulation/métamorphose de thrombusN16 [caillots] qui ont été déposés sur la paroi artérielle, puis incorporés à celle-ci. […]
[La maladie cardiovasculaire] se développe à la suite d’un dysfonctionnement dans un processus normal en trois étapes.
Première étape : détérioration de l’endothéliumN57/glycocalyxN80. Cette détérioration entraîne la deuxième étape : la formation d’un thrombus pour recouvrir la zone endommagée. Ce thrombus est à son tour recouvert d’une nouvelle couche d’endothélium qui attire efficacement le thrombus dans la paroi de l’artère.
Troisième étape : ce qui reste du thrombus est décomposé ou lysé par divers mécanismes de réparation, comme l’action des macrophages, qui peuvent décomposer et éliminer les restes de matériel endommagé.
Cependant, si les dommages sont accélérés, si les plaques sont plus grandes et/ou plus difficiles à décomposer, ou si la réparation est entravée, les plaques peuvent se développer et grossir. […]
Autrement dit, si le processus de détérioration se produit plus rapidement que la réparation, des plaques se formeront et grandiront. Si les systèmes de réparation fonctionnent à un rythme correspondant aux détériorations, la croissance des plaques sera ralentie, voire empêchée.
Ce concept s’appuie sur la publication Pourquoi l’athérothrombose est en principe une maladie hématologique (Sloop GD et al., 2018A50). Elspeth B Smith et W Douglas Thompson écrivaient avant eux (1994A51) :
Après avoir été négligé pendant de nombreuses années, le rôle de la thrombose dans l’infarctus du myocarde est réévalué. Il est de plus en plus évident que tous les aspects du système hémostatiqueN81 sont impliqués : non seulement dans l’événement occlusif aigu, mais aussi à tous les stades du développement de la plaque d’athérome, depuis l’initiation de l’athérogenèse jusqu’à l’expansion et la croissance des grandes plaques.
La perfusion d’activateur tissulaire recombinant du plasminogène (rt-PAN82) chez des hommes en bonne santé ne présentant aucun signe d’événements thrombotiques ou de conditions prédisposantes a entraîné une production significative de D‑dimèresN83, un fragment de fibrine réticulé […]. Ainsi, chez des sujets humains apparemment sains, il semble qu’une quantité importante de fibrine se dépose dans les artères, ce qui devrait donner à réfléchir sur la relation possible entre la coagulation et l’athérosclérose.
Selon Malcolm Kendrick (2022bA27 p. 429) :

Tous les vaisseaux sanguins sont tapissés de cellules endothéliales qui sont à leur tour recouvertes d’une couche de « gel », le glycocalyxN80, où l’oxyde nitrique (NO) est synthétisé, ainsi que de nombreux autres facteurs anticoagulants. Le glycocalyx agit comme une couche protectrice nécessaire pour maintenir la fonction et l’homéostasie des cellules endothéliales vasculaires. Il est également important de protéger les cellules endothéliales sous-jacentes des dommages physiques directs […].
Il est de plus en plus reconnu que dans de nombreuses maladies aiguës, le glycocalyx est aminci et fragilisé. Cela augmente à son tour le risque d’événements cardiovasculaires aigus […].
[…] Dans l’infection Sars-Cov2, le glycocalyx/endothélium est également attaqué et affaibli, et c’est ce qui déclenche les caillots sanguins que l’on observe fréquemment lors d’une infection par le coronavirus 2019 (COVID-19) [Vollenberg R et al., 2021A55].
Contrairement à la septicémie, où le glycocalyx est dégradé par un agent extérieur, dans le cas du syndrome respiratoire aigu sévère à coronavirus 2 (SARS-CoV2), les lésions se produisent à l’intérieur des cellules endothéliales. Les cellules endothéliales des poumons et du système vasculaire présentent une forte concentration de récepteurs de l’enzyme de conversion de l’angiotensine 2 [ACE2], et le SARS-CoV2 détourne ce récepteur pour pénétrer dans les cellules avant de se multiplier et d’éclater. Les cellules sont donc endommagées de l’intérieur, plutôt que de l’extérieur dans le cas d’une septicémie. Le résultat final est similaire.
[…] Si l’on considère les conditions à plus long terme, un facteur important connu pour causer des dommages chroniques au glycocalyx est un taux de glycémie élevé […].
Kendrick décrit d’autres conditions chroniques connues pour endommager le glycocalyxN80. Voir entre autres Jing Qu et al. (2021A39) et Alejandro Gomez Toledo et al. (2025N84). Le mécanisme est décrit ainsi (Kendrick 2022bA27 p. 430) :
Une fois l’endothélium endommagé, cela déclenche la formation d’un thrombusN16 [caillot]. L’un des mécanismes clés est qu’un endothélium sain et non endommagé exprime l’inhibiteur de la voie du facteur tissulaire (TFPIN85). Le TFPI limite l’action du facteur tissulaire (TFN86), qui est peut-être le facteur pro-coagulant le plus puissant. Ainsi, lorsque la production de TFPI diminue, le TF est libéré et entraîne la formation de thrombus [Neubauer K & B Zieger, 2022A34].
En plus de ce mécanisme, le glycocalyx est le lieu de synthèse du NO, un anticoagulant très puissant (en effet, il existe toute une série d’actions anticoagulantes qui nécessitent un glycocalyx sain pour fonctionner). Cela signifie qu’en cas de dommage, l’endothélium bascule vers un état prothrombotique
Une technique prometteuse pour l’évaluation de la dysfonction endothéliale serait la mesure de l’épaisseur du glycocalyx par des techniques d’imagerie microvasculaire, encore limitée à ce jour : « […] ces approches un peu naïves n’ont pas été confirmées à ce jour dans des études cliniques bien menées […] » (de Lorgeril M, 2022B4 p. 24–25).
Les facteurs qui interviennent dans les processus de formation et de destruction des thrombusN16 (caillots) sont le Facteur de von WillebrandN87, le fibrinogèneN88 et la lipoprotéine(a)N25 (Kendrick M, 2022bA27 p. 430).
L’auteur énonce ensuite les conditions de la destruction des thrombus qui se manifeste en même temps que leur formation (p. 431) :
Les dommages endothéliaux, la formation et la réparation des caillots représentent un processus continu. Ce phénomène est mis en évidence chez les personnes qui fument — et présentent un risque nettement plus élevé de maladie cardiovasculaire. Fumer entraîne directement des dommages au glycocalyx, une réduction de la production et de la biodisponibilité du NO qui, à son tour, crée un environnement pro-coagulant et inflammatoire. […] Cependant, le tabagisme stimule également la production et la libération de cellules progénitrices endothéliales (EPCN89) qui recouvrent les zones de lésions endothéliales, favorisant ainsi le processus de guérison […]. […]
Cela signifie que les dommages causés à l’endothélium/glycocalyx ne conduisent pas nécessairement à la formation (accélérée) de plaques d’athérosclérose.
Les EPC ne sont pas le seul dispositif de réparation. L’important, pour se protéger de la maladie cardiovasculaire, est d’assurer un bon équilibre entre les processus de dégradation et de réparation.
Dans son billet Pfizer and me – Best Buddies (Pfizer et moi, les meilleurs potes) (2022aA26), Malcolm Kendrick reproduit et commente une brochure titrée Pathological Triggers ‘New Insights into Cardiovascular Risk’ (Déclencheurs pathologiques « Nouvelles connaissances sur le risque cardiovasculaire ») qui avait été publiée par Pfizer en 1992, et dont le contenu soutenait intégralement l’hypothèse thrombogénique de l’athérosclérose. Kendrick commente :
Je dois préciser que c’était avant que Pfizer n’ait une statine. […] Puis, en 2000, Pfizer a racheté Warner Lambert, qui avait justement une statine appelée atorvastatine (Lipitor). Oui, Pfizer n’a pas réellement développé l’atorvastatine. Il a simplement racheté la société qui l’avait développée. Un joli coup.
Quoi qu’il en soit, en 1992, Pfizer n’était pas très intéressé par la réduction du cholestérol LDL, puisqu’il n’avait pas de statine. Cela signifie qu’ils avaient d’autres chats à fouetter avec leurs médicaments cardiovasculaires. Ils se concentraient davantage sur l’abaissement de la tension artérielle. Mais leur alpha-1-bloquant, la doxazocine (Cardura), n’était pas très efficace. […]
Malheureusement, en 1992, les médecins avaient déjà été lourdement et continuellement bombardés par le message selon lequel la réduction du cholestérol et de la tension artérielle était, de loin, la chose la plus importante à faire pour prévenir les maladies cardiovasculaires.
La coagulation du sang ? L’aspirine était utilisée, un peu. Mais c’était surtout pour contrer l’événement final : le gros caillot de sang qui bloque une artère principale. Personne n’a suggéré que la coagulation sanguine aurait quelque chose à voir avec l’athérosclérose elle-même.
Après tout, comment la coagulation du sang pourrait-elle être à l’origine du développement des plaques d’athérosclérose ? Comme on l’avait déjà dit à tout le monde, les plaques d’athérosclérose (les entités qui se développent progressivement et rétrécissent les artères) sont pleines de cholestérol. Pas des restes de caillots sanguins. […]
Cependant, ils ne sont pas allés très loin dans cette histoire [l’hypothèse thrombogénique]. Merck martelait la simvastatine (Zocor) et Bristol Myers Squibb promouvait également la pravastatine (Pravachol) avec une ferveur inébranlable. Le monde de la prévention des maladies cardiovasculaires s’orientait encore plus fermement vers la réduction du cholestérol et les statines. […]
À leur crédit (lucratif), ils [Pfizer] ont ensuite exceptionnellement bien surfé sur la vague du cholestérol.
⇪ L’hypothèse de prolifération tumorale
Cette hypothèse, avancée par les biologistes Earl Benditt (1977A5) et Russel Ross (1986A45), ne contredit pas l’hypothèse thrombogénique, mais elle peut expliquer certaines formations de plaques d’athérosclérose. Michel de Lorgeril la présente ainsi (2011B1 p. 89) :
Selon eux, les plaques d’athérosclérose sont dues à une prolifération tumorale de cellules musculaires dans des zones limitées des parois artérielles, prenant la forme de plaque plus ou moins obstructive. Cette prolifération serait une réponse à une altération fonctionnelle de l’endothélium.
Ici encore, le « mauvais cholestérol » LDL‑C n’est en rien responsable… L’hypothèse de prolifération tumorale pourrait expliquer la resténose après angioplastieN5, faisant suite au traumatisme subi par l’endothélium lors de cette intervention.
⇪ La lipoprotéine(a)
Le titre précédent « Mauvais cholestérol(s)» peut se décliner au pluriel parce qu’il existe une autre lipoprotéine, similaire au LDL, cette fois réellement nocive à la santé cardiovasculaire. Il s’agit de la lipoprotéine(a)N25 ou « Lp(a) » en abrégé. La molécule de Lp(a) se distingue de celle du LDL par le fait qu’elle est enveloppée dans de l’apolipoprotéine(a) ou « apo(a) » en abrégé (Kendrick M, 2021B6 p. 65–66).
➡ Attention de ne pas confondre l’apolipoprotéine(a) associée à la Lp(a) avec l’apolipoprotéine A1N37 qui fait partie du HDL…
L’industrie pharmaceutique a commencé à s’intéresser à la Lp(a) une fois qu’elle a cherché des médicaments qui permettraient d’en réduire le taux (Kendrick M, 2021B6 p. 284).
Fleur M. Van der Valk et al. (2016A53) ont identifié un mécanisme par lequel les lipoprotéines(a)N25 induiraient la migration de monocytesN90 dans la paroi artérielle, provoquant une réponse inflammatoire par l’intermédiaire de leur contenu en phospholipidesN91 oxydés. L’inflammation contribue à la formation de plaque et à sa fragilisation. Ils confirment ainsi que le taux de lipoprotéines(a) est un vrai facteur de risque cardiovasculaire, contrairement à celui du LDL‑C. Un taux élevé de Lp(a) est associé à un triplement du risque cardiovasculaire (Finneran P et al., 2021A17).
Les lipoprotéines(a)N25 bloquent la fibrinolyseN4 indispensable pour compenser la coagulation. En effet, l’apolipoprotéine(a) et le plasminogène, dont l’activation produit de la plasmineN92, ont des structures presque identiques qui ne se distinguent que par un « pli » différent à une extrémité. Cette différence du pli bloque l’activation du plasminogène (Kendrick M, 2021B6 p. 71).
Selon Kendrick (2022bA27 p. 431) :
Pourquoi la Lp(a) a‑t-elle cette fonction ? Linus Pauling a d’abord émis l’hypothèse que, comme l’homme ne peut pas synthétiser la vitamine C, qui joue un rôle clé dans la production de collagène, une carence en vitamine C entraînerait une dégradation et des « fissures » dans les parois des vaisseaux sanguins. La Lp(a), que l’on trouve presque exclusivement chez les animaux incapables de synthétiser la vitamine C, se lie très fortement à l’endothélium et à la paroi artérielle sous-jacente, créant des thrombusN16 particulièrement « résistants ». Cela réduit la perte de sang et permet à l’animal de survivre jusqu’à ce qu’il consomme suffisamment de vitamine C […]. Toutefois, ce rôle protecteur est une arme à double tranchant. En effet, la Lp(a) peut également favoriser la formation de thrombus qui sont plus difficiles à dissoudre. Cela entraîne à son tour une accélération de la formation de la plaque.
En résumé (Kendrick M, 2021B6 p. 65, 72) :
- Le nombre de molécules Lp(a) dans le sang est en général environ le quart ou le cinquième de celui des LDL, bien que certaines personnes aient plus de Lp(a) que de LDL. Ce niveau est déterminé génétiquement et tend à rester fixe. Aucun médicament n’a réussi à le diminuer significativement. Par conséquent, le diminuer ne rapporterait pas d’argent puisqu’il y a un silence presque assourdissant au sujet de la Lp(a).
- La structure des LDL et des Lp(a) est identique, à l’exception de l’attachement de la protéine apo(a) aux Lp(a).
- La Lp(a) est conçue pour protéger contre les lésions artérielles causées par une carence en vitamine C (et d’autres formes de lésions artérielles).
- La Lp(a) est incorporée dans les caillots sanguins qui se forment sur les parois artérielles endommagées.
- La Lp(a) rend les caillots sanguins beaucoup plus difficiles à éliminer.
- La Lp(a) peut se trouver en concentrations élevées dans les plaques d’athérosclérose.
- Un taux élevé de Lp(a) peut au moins tripler le risque de maladie cardiovasculaire.
La lipoprotéine(a) était un sujet d’étude de grand intérêt dans les années 1980 et au début des 1990, mais l’invention des statines a orienté la recherche dans une toute autre direction. Or. les statines ont très peu d’effet sur la Lp(a) ; elles ont au contraire tendance à en augmenter le taux… Michael B Boffa et Marlys S Koschinsky (2016A7) ont écrit :
Le moment est venu de réexaminer le potentiel prothrombotique/antifibrinolytique de la Lp(a) en vue de comprendre sa contribution mécanique aux événements athérothrombotiques.
Michael Eades a signalé dans The Arrow #139 qu’un médicament était en phase d’étude clinique, le premier capable de réduire le taux de Lp(a). Les industriels affirment :
Les médicaments courants qui réduisent le taux de LDL, comme les statines, n’ont pas le même effet sur le taux de Lp(a). En grande partie génétique, la Lp(a) est également difficile à contrôler par le régime alimentaire, l’exercice physique et d’autres changements de mode de vie.
Ces allégations sont inexactes : depuis plusieurs décennies, Eades et son épouse traitent avec succès des patients au taux élevé de Lp(a), en leur prescrivant un régime faible en glucides et riche en graisses saturées (LCHF). Leur expérience clinique a montré que le régime LCHF réduisait à la fois le taux de Lp(a) et la résistance à l’insulineN67, deux facteurs contribuant fortement aux problèmes cardiovasculaires.
L’action bénéfique des graisses saturées (d’origine animale) s’appuie sur plusieurs publications, parmi lesquelles Beverly A Clevidence et al. (1997N93) et Carra B Ebbeling et al. (2022N94). Pour ce qui concerne la première, Eades remarque :
Étant donné que l’objectif de l’étude était probablement de montrer que les graisses trans augmentent le taux de Lp(a) autant, sinon plus, que les graisses saturées, je suis sûr qu’ils ont été surpris de constater que les graisses saturées réduisaient de manière significative les taux de Lp(a).
Malcolm Kendrick recommande quant à lui (2021B6 p. 284) :
Que faire si vous avez un taux important de Lp(a) ? Je recommande de prendre de la vitamine B3 (niacineN95), si vous la tolérez, car elle peut provoquer des bouffées vasomotrices désagréables. Et un gramme par jour de vitamine C, plus environ 30 mg de co-enzyme Q10. De plus, je réduirais la consommation de glucides, surtout si j’ai un diabète de type 2. Je conseille aussi vivement de prendre de l’aspirine (75 mg par jour) pour éviter la formation de caillots.
Je ne suivrais pas sa recommandation d’aspirine, vue sa toxicité, entre autres l’augmentation de risque de DMLAN96 (Kahawita SK et al., 2015A24). Sa toxicité sur le long terme est décrite et documentée par Michel de Lorgeril (2022B4 p. 68–70). D’autres antiagrégants plaquettairesN97 existent — voir mon article Soigner ses artères. Peut-être pas non plus les vitamines qui ne remplacent pas un rééquilibrage nutritionnel — voir ma page Compléments alimentaires.
⇪ Donc ne pas faire de régime ?
Il ne faudrait pas conclure hâtivement qu’aucun régime alimentaire n’est susceptible de diminuer le risque cardiovasculaire. Certains régimes ont une incidence bénéfique mesurable, mais ce que démontrent les travaux cités est que cette incidence n’est pas la conséquence d’une diminution du taux de cholestérol.
Dave Feldman — qui n’est pas médecin mais ingénieur en informatique — a effectué des mesures de l’effet sur les taux sanguins de LDL‑C de la consommation de glucides et lipides (voir son siteN98), montrant que ce taux peut varier fortement en quelques jours selon ce qui a été ingéré. Cette variabilité est utilisée par les cardiologues — à la manière des traders — pour prescrire des statines lorsque le taux affiche une augmentation…
Une des publications les plus souvent citées sur la réduction du risque cardiovasculaire associée à une meilleure hygiène alimentaire est l’Étude de Lyon (de Lorgeril M et al., 1999A12) qui consistait à suivre, sur une durée moyenne de 46 mois, des patients déjà victimes d’un accident cardiovasculaire, à qui l’on avait prescrit des mesures de prévention secondaire pour éviter une récidive. Cette étude a montré les effets salutaires d’une diète méditerranéenneN99 en comparaison avec les habitudes alimentaires antérieures de ces sujets. Toutefois, une étude interventionnelle a montré ultérieurement « qu’un régime pauvre en glucides pourrait être plus efficace qu’un régime méditerranéen pour favoriser la perte de poids et améliorer divers facteurs de risque métaboliques et cardiovasculaires chez les patients en surpoids/obèses atteints de diabète dee type 2 » (Currenti W et al., 2023N100).
D’autres travaux ont confirmé qu’une partie significative de l’effet protecteur d’un tel régime était un ratio oméga 3 sur oméga 6 plus élevé. En effet, la formation de plaque d’athéroscléroseN2 est fortement conditionnée par un mécanisme d’inflammation de l’épithéliumN54 des artères, indépendamment de la présence de lipoprotéinesN20, sous l’effet de contraintes mécaniques (Hallow KM et al., 2009A19). Or les oméga 6 augmentent cette inflammation alors que les oméga 3 la réduisent.
Alors que les Français consomment plus de graisses saturées que les habitants d’autres pays, leur taux de maladies coronariennes est moins élevé (French paradoxN33). Par exemple, au Royaume-Uni on consomme environ 13.5 % des calories sous forme de graisses saturées, contre 15.5 % en France, alors que le taux de décès par maladies de cœur est seulement de 22 pour 100 000 en France contre 63 pour 100 000 au Royaume-Uni.
L’American Heart Association recommande de limiter la consommation de graisses saturées à moins de 7 % du total des calories. Or la Lituanie, avec son taux moyen de 7.7 %, est un des pays à plus forte mortalité cardiaque : 122 pour 100 000 — cité par J. MercolaN101.
Le French paradox a trouvé une explication dans de nombreuses études établissant que la consommation de graisses saturées n’est pas un facteur de risque de diabète de type 2N102 ni de maladies cardiovasculaires. Voir par exemple la méta-analyse publiée en 2015 par De Souza, RJ et al.A13 dans le British Medical Journal. Les graisses hydrogénées (acides gras transN103) — margarines et autres préparations industrielles — sont les principales responsables de ces pathologies.
La méta-analyse de Siri-Tarino PW et al., 2010A49 conclut à l’absence de preuve d’un lien entre la consommation de graisses saturées et une maladie coronarienne, y compris les accidents cardiovasculaires. Blekkenhorst LC et al. (2015A6) résument :
Bien qu’il y ait une forte association positive entre l’apport en acides gras saturés et le cholestérol LDL, le cholestérol LDL n’était pas associé à la mortalité par maladie cardiovasculaire dans cette cohorte.
Ce qui ne les empêche pas d’ajouter : « Néanmoins, ces données soutiennent les conseils diététiques pour réduire l’apport en acides gras saturés. »
Contrairement à une croyance répandue chez les adeptes de régimes riches en graisse (et pauvres en glucides), la consommation de graisses saturées peut augmenter le taux de cholestérol LDL. Marit Kolby Zinöcker et collègues expliquent cela dans le contexte de la biologie adaptative humaine (Zinöcker MK et al., 2021N104) :
[…] Nous proposons un nouveau modèle, le modèle d’adaptation homéoviscérale aux lipides alimentaires (HADL), qui explique les variations du cholestérol des lipoprotéines comme des ajustements homéostatiques adaptatifs servant à maintenir la fluidité de la membrane cellulaire et, par conséquent, une fonction cellulaire optimale. En raison de l’apport très variable d’acides gras chez l’homme et les autres espèces omnivores, nous proposons que les lipoprotéines circulantes servent de tampon pour permettre la redistribution rapide des molécules de cholestérol entre les cellules et les tissus spécifiques, qui est nécessaire en cas de changements dans l’apport d’acides gras alimentaires. Par conséquent, les taux circulants de cholestérol LDL peuvent changer pour des raisons non pathologiques.
Une étude observationnelle (Jakobsen MU et al., 2009A22) couvrant 344 000 personnes pendant six ans suggère même que le remplacement de graisses saturées par des glucides ou des graisses mono-insaturées (huiles végétales) augmenterait le risque de maladie coronarienne.
Des équipes en lien d’intérêt avec l’industrie des statines s’efforcent de ressusciter la croyance en une relation causale entre consommation d’aliments riches en cholestérol et maladies cardiovasculaires. C’est le cas de Zhong, VW et al. (2019A61) déclarant, à partir d’une méta-analyse de 6 études observationnelles, que la consommation d’œufs serait associée en relation « dose-dépendante » à un risque accru de maladie cardiovasculaire… Leur méta-analyse ne remplit pas les critères de HillN105 permettant d’inférer une causalité à partir d’une simple corrélation. D’autre part, les personnes rapportant le plus faible apport en cholestérol alimentaire avaient également un apport énergétique significativement plus faible : un tiers seulement de l’apport énergétique de celles dont l’apport en cholestérol était le plus élevé.
Zoë Harcombe suggère (2019N106) : « Peut-être que les gens ne mangeaient pas plus d’œufs ou de cholestérol alimentaire – ils étaient juste plus honnêtes au sujet de leur consommation de nourriture, ou mieux à même s’en souvenir ! » Ce biais est caractéristique de la collecte de données « basées sur la mémoire » décrite dans mon article Faut‐il jeter les enquêtes nutritionnelles ? D’autres biais ont été soulignés, comme par exemple le fait que les données nutritionnelles ont été collectées une fois pour toutes au début de la période de 17 ans d’observation — comme si les habitudes alimentaires étaient restées identiques. Enfin, il ne s’agissait pas seulement d’œufs mais d’aliments contenant des œufs, ce qui inclut, chez des consommateurs nord-américains, de nombreux produits transformés : crèmes glacées, gâteaux etc.
➡ Les biais de collecte de données dans les études observationnelles peuvent aussi affecter la plupart des études citées sur cette page. Leurs conclusions doivent donc être lues avec un esprit critique et confrontées à celles d’autres travaux.
La consommation de produits laitiers (lait, fromage, yaourt) a été associée à une réduction de la mortalité et du risque cardiovasculaire dans l’étude de cohorte multicentrique PURE pendant 9 ans sur 18 pays de 5 continents (2017N107) :
Un apport plus élevé de produits laitiers totaux (> 2 portions par jour comparé à l’absence de prise) était associé à un risque moindre de résultat composite (HR 0,84, IC à 95 %: 0,75 à 0,94 ; p = 0,0004), mortalité totale (0,83, 0,72–0,96 ; p = 0,0052), mortalité non cardiovasculaire (0,86, 0,72 à 1,02 ; p = 0,046), mortalité cardiovasculaire (0,77, 0,58–1,01 ; p = 0,029), maladies cardiovasculaires majeures (0,78, 0,67–0,90 ; p = 0,0001) et AVC (0,66, 0,53–0,82 ; p = 0,0003).
Pour ce qui concerne le risque de maladie coronarienne, la méta-analyse de Mozaffarian D et al. (2011A32) classe en détail les influences de divers aliments. Une description claire du régime méditerranéen popularisé par l’Étude de Lyon est présentée dans l’ouvrage Prévenir l’infarctus et l’accident vasculaire cérébral (de Lorgeril M, 2011B1).
De nombreux liens vers des articles scientifiques et commentaires (en anglais) au sujet du cholestérol et des traitements qui lui sont associés se trouvent sur une page du site The International Network of Cholesterol SkepticsC9.
Il n’est pas anodin de signaler que la préconisation de remplacer les graisses saturées d’origine animale par des huiles végétales insaturées figure encore dans les recommandations de santé publique de nombreux pays, dont la France. La Suède a été le premier pays à effectuer un revirement total en 2013C8… En 2015C10, le Gouvernement des USA a décidé de retirer sa mise en garde contre le cholestérol alimentaire, qui était en vigueur depuis près de 40 ans. (Voir les Dietary Guidelines for Americans 2015–2020B5.)
Parmi les explications plausibles de la persistence de recommandations inadéquates, le compte-rendu de Kearns CE et al. (2016A25) révèle que la Sugar Research FoundationN108 a manipulé les chercheurs pour qu’ils dissimulent le lien causal entre la consommation de sucre et les maladies cardiovasculaires. Le cholestérol a été désigné comme ennemi numéro 1 de la santé cardiovasculaire à une époque où les industries du tabac et de la production sucrière prenaient un essor exceptionnel aux USA.
⇪ Quid des médicaments ?
J’entends souvent dire que s’il est admis que « faire du régime » ne convient pas à tout le monde, par contre il est depuis longtemps affirmé que le contrôle médicamental des taux de cholestérol diminuerait significativement les risques d’accident cardiovasculaire et d’AVC ischémique. C’est le sujet de mon article Statines et médicaments anticholestérol.
Indubitablement, les statines et autres médicaments, comme certains régimes « anti-cholestérol », font baisser le taux de LDL‑C, mais nous avons vu que l’existence d’un lien de causalité entre une baisse du cholestérol et une diminution du risque cardiovasculaire avait été remise en cause. Mon article montre aussi que les effets indésirables de ces médicaments ont été délibérément ignorés ou sous-évalués.
⇪ Références
⇪ ✓ Articles
🔵 Notes pour la version papier :
- Les identifiants de liens permettent d’atteindre facilement les pages web auxquelles ils font référence.
- Pour visiter « 0bim », entrer dans un navigateur l’adresse « https://leti.lt/0bim ».
- On peut aussi consulter le serveur de liens https://leti.lt/liens et la liste des pages cibles https://leti.lt/liste.
- A1 · o4hz · A Midwestern Doctor (2025). What They Don’t Tell Us About Heart Disease. Site The Forgotten Side of Medicine.
- A2 · ym4r · Ahmadi, SA et al. (2008). The impact of low serum triglyceride on LDL-cholesterol estimation. Arch Iran Med. 11, 3 : 318–321.
- A3 · 0402 · Balasubramaniam, S et al. (1994). Circadian rhythm in hepatic low-density-lipoprotein (LDL)-receptor expression and plasma LDL levels. Biochem J., 298 : 39–43.
- A4 · hta9 · Banta, JE et al. (2018). The Global Influence of the Seventh-Day Adventist Church on Diet. Religions 9, 9 : 251.
- A5 · b1pi · Benditt, EP (1977). The Origin of Atherosclerosis. Scientific American Magazine 236, 2 : 74.
- A6 · 6ran · Blekkenhorst, LC et al. (2015). Dietary saturated fat intake and atherosclerotic vascular disease mortality in elderly women : a prospective cohort study. American Journal of Clinical Nutrition 101, 6 : 1263–1268. doi:10.3945/ajcn.114.102392
- A7 · iap7 · Boffa, MB & MS Koschinsky (2016). Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease ? J Lipid Res, 57, 5:745–757.
- A8 · y0t2 · Bray, MS et al. (2011). Regulation of Fatty Acid Metabolism by Cell Autonomous Circadian Clocks : Time to Fatten up on Information ? The Journal of Biological Chemistry, 286 : 11883–11889.
- A9 · vbtx · Chowdhury, R et al. (2014). Association of Dietary, Circulating, and Supplement Fatty Acids With Coronary Risk : A Systematic Review and Meta-analysis. Annals of Internal Medicine, 160, 6 : 398–406.
- A10 · ih32 · Davidson, MH et al. (2011). Clinical utility of inflammatory markers and advanced lipoprotein testing : advice from an expert panel of lipid specialists. J. Clin. Lipidol., 5, 5 : 338–367.
- A11 · qexb · Dayton, S. et al. (1969). A Controlled Clinical Trial of a Diet High in Unsaturated Fat in Preventing Complications of Atherosclerosis. Circulation, July, 40, 1S2.
- A12 · gqpv · de Lorgeril M ; Salen P ; Martin JL ; Monjaud I ; Delaye J ; Mamelle N (1999). Mediterranean Diet, Traditional Risk Factors, and the Rate of Cardiovascular Complications After Myocardial Infarction. Circulation, 99 : 779–785.
- A13 · ow45 · De Souza, RJ et al. (2015). Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes : systematic review and meta-analysis of observational studies. BMJ 2015 ; 351.
- A14 · ys9f · DiNicolantonio, JJ & JH O’Keefe (2018). Omega‑6 vegetable oils as a driver of coronary heart disease : the oxidized linoleic acid hypothesis. Open Heart 5 : e000898.
- A15 · gjl6 · DuBroff, R (2018). A Reappraisal of the Lipid Hypothesis. The American Journal of Medicine 131, 9 : 993–997.
- A16 · 4bsh · Duval, S (2013). Cholestérol : le bon, le mauvais et les truands. Site du Formindep.
- A17 · ic85 · Finneran, P et al. (2021). Lipoprotein(a) and Coronary Artery Disease Risk Without a Family History of Heart Disease. J Am Heart Assoc. 10, 5 : e017470.
- A18 · k913 · Gertz, SD et al. (1991). Composition of atherosclerotic plaques in the four major epicardial coronary arteries in patients greater than or equal to 90 years of age. Am J Cardiol. 67, 15 : 1228–1233.
- A19 · x9fh · Hallow, KM ; Taylor, WR ; Rachev, A.; Vito, RP (2009). Markers of inflammation collocate with increased wall stress in human coronary arterial plaque. Biomechanics and Modeling in Mechanobiology, December, 8, 6 : 473–486.
- A20 · lj25 · Heir, T et al. (2016). Cholesterol and prostate cancer risk : a long-term prospective cohort study. BMC Cancer, August, 16 : 643.
- A21 · e1nk · Hu, P et al. (2020). Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease : A Systematic Review and Meta-Analysis. Circulation 141, 22 : 1742–1759.
- A22 · r32n · Jakobsen, MU et al. (2009). Major types of dietary fat and risk of coronary heart disease : a pooled analysis of 11 cohort studies. Am J Clin Nutr, 89, 5 : 1425–32.
- A23 · pcr1 · Johansson, I et al. (2012). Associations among 25-year trends in diet, cholesterol and BMI from 140,000 observations in men and women in Northern Sweden. Nutrition Journal, 11, 40.
- A24 · cgr5 · Kahawita, SK et al. (2015). Aspirin use and early age-related macular degeneration : a meta-analysis (reprint). Canadian Journal of Ophthalmology 50, suppl 1 : S29-S33.
- A25 · e4mt · Kearns, CE et al. (2016). Sugar Industry and Coronary Heart Disease Research : A Historical Analysis of Internal Industry Documents. JAMA Intern Med., 176, 11 : 1680–1685.
- A26 · nm7g · Kendrick, M (2022a). Pfizer and me – Best Buddies. Site du Dr. Malcolm Kendrick, 21 mars.
- A27 · l92n · Kendrick, M (2022b). Assessing cardiovascular disease : looking beyond cholesterol. Current Opinion in Endocrinology & Diabetes and Obesity : August 18. doi:10.1097/MED.0000000000000761
- A28 · es06 · Kresser, C (2013). The Diet-Heart Myth : Why Everyone Should Know Their LDL Particle Number. Site Let’s take back our health.
- A29 · py6s · Kumi, D et al (2024). Current US prevalence of myocardial injury patterns and clinical outcomes among hospitalised patients with familial hypercholesterolaemia : insight from the National Inpatient Sample—a retrospective cohort study BMJ Open 14 : e077839.
- A30 · vk02 · Li, D & JL Mehta (2005). Oxidized LDL, a critical factor in atherogenesis. Cardiovascular Research, 68, 3 : 353–354. doi:10.1016/j.cardiores.2005.09.009
- A31 · pmt3 · Minelli, S et al. (2020). Reflections on Atherosclerosis : Lesson from the Past and Future Research Directions. J Multidiscip Healthc, 13 : 621–633. doi:10.2147/JMDH.S254016
- A32 · m74m · Mozaffarian, D et al. (2011). Components of a Cardioprotective Diet. Circulation, 123, 24.
- A33 · k55q · Multiple Risk Factor Intervention Trial Research Group (1982). Multiple risk factor intervention trial. Risk factor changes and mortality results. JAMA Sept 24, 248, 12:1465–77.
- A34 · qz1n · Neubauer, K & B Zieger (2022). Endothelial cells and coagulation. Cell Tissue Res. 387, 3:391–398.
- A35 · k1f0 · Nguyen, XMT et al. (2023). Serum Cholesterol and Impact of Age on Coronary Heart Disease Death in More Than 4 Million Veterans. Journal of the American Heart Association 12, 21 : e030496.
- A36 · i8kv · Paddock, C (2017). High cholesterol diagnosis tied to lower breast cancer risk. Medical News Today, on-line journal.
- A37 · 71f0 · Petursson, H et al. (2012). Is the use of cholesterol in mortality risk algorithms in clinical guidelines valid ? Ten years prospective data from the Norwegian HUNT 2 study. J Eval Clin Pract., 18, 1 : 159–168.
- A38 · jzti · Potluri, R et al. (2017) . Patients with a diagnosis of hyperlipidaemia have a reduced risk of developing breast cancer and lower mortality rates : a large retrospective longitudinal cohort study from the UK ACALM registry. European Heart Journal, 38, suppl_1, August, ehx504.3106. doi:10.1093/eurheartj/ehx504.3106.
- A39 · ez47 · Qu, J et al. (2021). Glycocalyx Impairment in Vascular Disease : Focus on Inflammation. Front. Cell Dev. Biol. 9 : 730621.
- A40 · xvn9 · Ramsden, CE et al. (2013). The Sydney Diet Heart Study : a randomised controlled trial of linoleic acid for secondary prevention of coronary heart disease and death. FASEB Journal 27, 51 : 127. doi:10.1096/fasebj.27.1_supplement.127.4
- A41 · cy0i · Ramsden, CE et al. (2016). Re-evaluation of the traditional diet-heart hypothesis : analysis of recovered data from Minnesota Coronary Experiment (1968–73). BMJ, 353 : i1246.
- A42 · 9fur · Ravnskov, U (2003). High cholesterol may protect against infections and atherosclerosis. QJM : An International Journal of Medicine, 96, 12 : 927–934.
- A43 · z8ku · Ravnskov, U et al. (2016). Lack of an association or an inverse association between low-density-lipoprotein cholesterol and mortality in the elderly : a systematic review. BMJ Open, 6:e010401. doi:10.1136/bmjopen-2015–010401
- A44 · n25q · Reddy, VS et al. (2015). Relationship Between Serum Low-Density Lipoprotein Cholesterol and In-hospital Mortality Following Acute Myocardial Infarction (The Lipid Paradox). American Journal of Cardiology, 115, 5:557–562.
- A45 · wq6q · Ross, R (1986). The pathogenesis of atherosclerosis – an update. N Engl J Med 314, 8 : 488–500.
- A46 · hb2e · Rothberg, MB (2013). Coronary Artery Disease as Clogged Pipes – A Misconceptual Model. Circulation : Cardiovascular Quality and Outcomes, 6:129–132.
- A47 · b3zc · Rozanski, A et al. (2022). Association between hypercholesterolemia and mortality risk among patients referred for cardiac imaging test : Evidence of a “cholesterol paradox?” Progress in Cardiovascular Diseases, in press. doi:10.1016/j.pcad.2022.10.007
- A48 · e2yy · Sachdeva, A et al. (2009). Lipid levels in patients hospitalized with coronary artery disease : An analysis of 136,905 hospitalizations in Get With The Guidelines. American Heart Journal, 157, 1 : 111–117.
- A49 · mp15 · Siri-Tarino, PW et al. (2010). Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. Am J Clin Nutr, 91, 3 : 535–546.
- A50 · f9y4 · Sloop, GD et al. (2018). Why Atherothrombosis is in Principle a Hematologic Disease : The Effect of Disorders and Drugs which Affect Thrombosis on the Development of Atherosclerotic Plaques. Int Arch Cardiovasc Dis 2:012. doi:10.23937/IACVD-2017/1710012
- A51 · wk11 · Smith, EB & WD Thompson (1994). Fibrin as a factor in atherogenesis. is, Thrombosis Research 73, 1 : 1–19.
- A52 · e5a3 · Tedgi, A & Z Mallat (2006). Cytokines in Atherosclerosis : Pathogenic and Regulatory Pathways. Physiol Rev, April, 86, 2. doi:10.1152/physrev.00024.2005
- A53 · b6pw · van der Valk, FM et al. (2016). Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation, 134, 8 : 611–624.
- A54 · t01t · Velican, C & D Velican (1983). Progression of coronary atherosclerosis from adolescents to mature adults. Athrosclerosis 47, 2 : 131–144.
- A55 · yi5d · Vollenberg, R et al. (2021). Indications of Persistent Glycocalyx Damage in Convalescent COVID-19 Patients : A Prospective Multicenter Study and Hypothesis. Viruses 3, 11 : 2324.
- A56 · vclk · Wang, DW et al. (2016). Association of Specific Dietary Fats With Total and Cause-Specific Mortality. JAMA Intern Med. 2016;176(8):1134–1145. doi:10.1001/jamainternmed.2016.2417
- A57 · b1te · Wang, TY et al. (2001). Low Triglyceride Levels Affect Calculation of Low-Density Lipoprotein Cholesterol Values. Arch Pathol Lab Med 125, 3 : 404–405.
- A58 · u3vk · Ward, T (2018). HDL‑C : is it time to stop calling it the ‘good’ cholesterol ? Medscape/Disclosures, July.
- A59 · 1t5f · Wilkins, JT et al. (2016). Discordance Between Apolipoprotein B and LDL-Cholesterol in Young Adults Predicts Coronary Artery Calcification. Journal of the American College of Cardiology, 67, 2 : 193–201.
- A60 · 39n0 · Yusuf, S et al. (2004). Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. The Lancet, 364, 9438 : 937–952.
- A61 · b78q · Zhong, VW et al. (2019). Associations of Dietary Cholesterol or Egg Consumption With Incident Cardiovascular Disease and Mortality. JAMA, 321, 11:1081–1095. doi:10.1001/jama.2019.1572.
⇪ ✓ Ouvrages et rapports
- B1 · ad6o · De Lorgeril, M (2011). Prévenir l’infarctus et l’accident vasculaire cérébral. Thierry Souccar.
- B2 · 8wtg · De Lorgeril, M (2013). Cholestérol, mensonges et propagande. Thierry Souccar, 2e édition. ➡ Éviter la version Kindle qui est mal codée.
- B3 · 588e · De Lorgeril, M (2015). L’horrible vérité sur les médicaments anti cholestérol – Comment les statines empoisonnent en silence. Thierry Souccar. ➡ Éviter la version Kindle qui est mal codée.
- B4 · rm72 · de Lorgeril, M (2022). Comment échapper à l’infarctus et l’AVC. Thierry Souccar.
- B5 · 0wn9 · Health.gov (2015). Dietary Guidelines for Americans 2015–2020
- B6 · j28q · Kendrick M (2021). The Clot Thickens. Columbus Publishing.
- B7 · q5f8 · Taubes, G (2007). Good Calories, Bad Calories : Challenging the Conventional Wisdom on Diet, Weight Control, and Disease. Knopf.
⇪ ✓ Divers
- C1 · wn8z · Bailey, R (2016). Santé : aucun risque à manger de la viande, du fromage, du beurre.
- C2 · pm34 · De Lorgeril, M (2024). Infarctus du myocarde, mortalité et Hypercholestérolémie Familiale. Blog personnel.
- C3 · y9ft · Dufournet, B (2022). Bilan lipidique (cholestérol, triglycérides): interprétation ! Vidéo YouTube.
- C4 · ex2f · Dufournet, B (2023). Le LDL n’est pas votre ennemi ! Pourquoi les médecins sont-ils mal informés ? Vidéo YouTube.
- C5 · t6fq · Ennezat, P‑V (2018). Vidéo “Le cholestérol”. Congrès Médecine Générale France. Paris : 5–7 avril. Vidéo sur YouTube.
- C6 · zw96 · Kendrick, M (2023). Losing Faith In Medical Research. Lecture at Broken Science Phoenix Event. Video recording.
- C7 · 1g8x · Raton, P (2017). 1001 raisons de ne pas faire baisser son cholestérol à l’aide de médicaments hypocholestérolémiants. Blog.
- C8 · 0s5o · Shilhavy, Brian (2017). Sweden Becomes First Western Nation to Reject Low-fat Diet Dogma in Favor of Low-carb High-fat Nutrition.
- C9 · n5mv · The International Network of Cholesterol Skeptics (THINCS). About Cholesterol and its Lowering.
- C10 · 108r · Whoriskey, Peter (2015). The U.S. government is poised to withdraw longstanding warnings about cholesterol.
⇪ ▷ Liens
- N1 · br8z · Syndrome coronarien aigu – Wikipedia
- N2 · mnd6 · Athérome ou athérosclérose – Wikipedia
- N3 · 5m77 · Infarctus du myocarde – Wikipedia
- N4 · xdqj · Fibrinolyse – Wikipedia
- N5 · 55c4 · Angioplastie coronaire – Wikipedia
- N6 · eb7u · Stent – Wikipedia
- N7 · dh7o · Pontage aorto-coronarien – Wikipedia
- N8 · jrzg · Accident vasculaire cérébral – AVC – Wikipedia
- N9 · q6ty · Hémorragie – Wikipedia
- N10 · f42s · Ischémie – Wikipedia
- N11 · t9th · Échographie Doppler – Wikipedia
- N12 · ty6z · Angiographie – Wikipedia
- N13 · 5z51 · Scintigraphie – Wikipedia
- N14 · se8i · Épreuve d’effort – Wikipedia
- N15 · v2hy · Paresthésie – Wikipedia
- N16 · tgae · Thrombus – Wikipedia
- N17 · ns68 · Vidéo “Coronaires : retrouvez la lumière”
- N18 · a9oi · Fibrome – Wikipedia
- N19 · r2k7 · Coronary Artery Calcification – From Mechanism to Molecular Imaging
- N20 · d55y · Lipoprotéine – Wikipedia
- N21 · 2ss4 · Lipoprotéine de haute densité – HDL – Wikipedia
- N22 · sulv · Lipoprotéine de basse densité – LDL – Wikipedia
- N23 · 1edl · Triglycéride – Wikipedia
- N24 · 8wrb · Lipoprotéine de très basse densité – VLDL – Wikipedia
- N25 · xxly · Lipoprotéine(a) – Wikipedia
- N26 · w3a0 · Rythme nycthéméral – Wikipedia
- N27 · 23mb · Statine – Wikipedia
- N28 · jxc5 · Simvastatin with or without Ezetimibe in Familial Hypercholesterolemia
- N29 · 18ym · Étude de Framingham – Wikipedia
- N30 · 8gis · Framingham Risk Score – Wikipedia
- N31 · o7g1 · Ancel Keys – Wikipedia
- N32 · wgy2 · Seven Countries Study – Wikipedia
- N33 · p6yd · The French paradox : lessons for other countries
- N34 · 3qaz · Serge Renaud – Wikipedia
- N35 · avx0 · Video “An Update on Demonization and Deception in Research on Saturated Fat” : The French paradox – David M. Diamond
- N36 · gkgi · Apolipoprotéine B – Wikipedia
- N37 · jxne · Apolipoprotéine A1 – Wikipedia
- N38 · 0y9v · Oméga‑6 – Wikipedia
- N39 · kqzi · Oméga‑3 – Wikipedia
- N40 · lcub · Essai randomisé contrôlé – Wikipedia
- N41 · cvs9 · Vidéo “C’est le gras qui bouche les artères ? Un cardiologue répond” – Pierre-Vladimir Ennezat
- N42 · f295 · Errigo, A et al. (2025). The Cholesterol Paradox in Long-Livers from a Sardinia Longevity Hot Spot (Blue Zone). Nutrients 17, 5 : 765.
- N43 · 28he · Frank Hu
- N44 · piaz · Association of Specific Dietary Fats With Total and Cause-Specific Mortality – PubPeer comments
- N45 · 9jo9 · California Walnut Commission
- N46 · wo61 · Régression de Cox – Wikipedia
- N47 · qanw · Hypercholestérolémie familiale – Wikipedia
- N48 · mdjj · Estimated total deaths by cause and WHO Member State – XLS file
- N49 · voz9 · Total cholesterol levels versus mortality data from 164 countries – Ricardo Carvalho
- N50 · yf5o · Bartelt, K et al. (2024). Low LDL and HDL Correlated with Increased Likelihood of Myocardial Infarction and Coronary Artery Disease. Epic Research, Dual-Team Study.
- N51 · kow3 · Video “Dr Aseem Malhotra” – The Joe Rogan Experience
- N52 · cv9w · Michel de Lorgeril – Wikipedia (archive)
- N53 · lda0 · Glycosylation – Wikipedia
- N54 · 2p75 · Épithélium – Wikipedia
- N55 · kqvd · L’inflammation de bas grade ou inflammation systémique
- N56 · ua5u · Revascularization – Wikipedia
- N57 · pgyc · Endothélium – Wikipedia
- N58 · oq6b · Sténose – Wikipedia
- N59 · ifem · Angine de poitrine – Wikipedia
- N60 · 60vw · Macrophage – Wikipedia
- N61 · qpca · Mastocyte – Wikipedia
- N62 · 8464 · Autoimmunity, autoantigen – Wikipedia
- N63 · fufq · Cytokine – Wikipedia
- N64 · bcha · Peptidase, protéase – Wikipedia
- N65 · clpm · Estimation of LDL particles via cholesterol content – Wikipedia
- N66 · th45 · Video “An Update on Demonization and Deception in Research on Saturated Fat” – David Diamond
- N67 · dlmy · Résistance à l’insuline – Wikipedia
- N68 · kpej · Syndrome métabolique – Wikipedia
- N69 · dzdm · Leaky gut syndrome – Wikipedia
- N70 · upe3 · Cellule spumeuse – Wikipedia
- N71 · k90q · Érythrocyte – Wikipedia
- N72 · mob9 · Jonction serrée – Wikipedia
- N73 · tnk4 · Vasa vasorum – Wikipedia
- N74 · v1ja · Fatty streak – Wikipedia
- N75 · bc5m · Ester – Wikipedia
- N76 · zbu3 · Rath, M et al. (1989). Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. ATVB 9, 5 : 579–592.
- N77 · i0s9 · Prostacycline – Wikipedia
- N78 · yqv1 · Acide linoléique – Wikipedia
- N79 · ntr4 · Thrombogenicity – Wikipedia
- N80 · sei1 · Glycocalyx – Wikipedia
- N81 · v9d3 · Hémostase – Wikipedia
- N82 · g5i2 · Activateur tissulaire du plasminogène tPA – Wikipedia
- N83 · bcx8 · D‑dimère – Wikipedia
- N84 · a9bz · Gomez Toledo, A et al. (2025).Endothelial Glycocalyx Turnover in Vascular Health and Disease : Rethinking Endothelial Dysfunction. Annual Review of Biochemistry 94 : 561–586.
- N85 · bf1v · Tissue factor pathway inhibitor (TFPI) – Wikipedia
- N86 · z159 · Facteur tissulaire – Wikipedia
- N87 · vr3m · Facteur de von Willebrand – Wikipedia
- N88 · n9kt · Fibrinogène – Wikipedia
- N89 · o4po · Endothelial progenitor cell – Wikipedia
- N90 · 8jao · Monocyte – Wikipedia
- N91 · p7iw · Phospholipide – Wikipedia
- N92 · kl1s · Plasmine – Wikipedia
- N93 · swo9 · Plasma Lipoprotein (a) Levels in Men and Women Consuming Diets Enriched in Saturated, Cis‑, or Trans-Monounsaturated Fatty Acids
- N94 · sl82 · Effects of a low-carbohydrate diet on insulin-resistant dyslipoproteinemia—a randomized controlled feeding trial
- N95 · nq3x · Vitamine B3 – Wikipedia
- N96 · fij6 · Dégénérescence maculaire liée à l’âge (DMLA) – Wikipedia
- N97 · 0vhz · Antiagrégant plaquettaire – Wikipedia
- N98 · ux2z · Site “Cholesterol Code – Reverse Engineering the Mystery” – Dave Feldman
- N99 · 6edf · Régime méditerranéen – Wikipedia
- N100 · mjy0 · Currenti, W et al. (2023). Comparative Evaluation of a Low-Carbohydrate Diet and a Mediterranean Diet in Overweight/Obese Patients with Type 2 Diabetes Mellitus : A 16-Week Intervention Study. Nutrients 16, 1 : 95.
- N101 · 37ee · Statin Nation II : What Really Causes Heart Disease ?
- N102 · a3u9 · Diabète de type 2 – Wikipedia
- N103 · q7py · Acide gras trans – Wikipedia
- N104 · oeu0 · Zinöcker, MK et al. (2021). The homeoviscous adaptation to dietary lipids (HADL) model explains controversies over saturated fat, cholesterol, and cardiovascular disease risk. The American Journal of Clinical Nutrition 113,2 : 277–289.
- N105 · d2q1 · Hill’s Criteria for Causality
- N106 · knvx · Eggs, cholesterol & CVD
- N107 · ejg5 · Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): a prospective cohort study
- N108 · 4stf · Sugar Research Foundation, Inc.
Article créé le 30/01/2017 - modifié le 8/06/2026 à 15h52 • 11 138 visites



{kind=link}